Home » Student Resources » Online Chemistry Textbooks » CH450 and CH451: Biochemistry - Defining Life at the Molecular Level » Chapter 9: DNA Replication
MenuCH450 and CH451: Biochemistry - Defining Life at the Molecular Level
Chapter 9: DNA Replication
9.1 DNA Replication is Semiconservative
9.2 DNA Replication in Prokaryotes
9.3 DNA Replication of Extrachromosomal Elements: Plasmids and Viruses
9.4 DNA Replication in Eukaryotes
9.5 Replication of Mitochondrial DNA
9.6 Telomeres and Replicative Senescence
9.7 References
9.1 DNA Replication is Semiconservative
The elucidation of the structure of the double helix by James Watson and Francis Crick in 1953 provided a hint as to how DNA is copied during the process of DNA replication. Separating the strands of the double helix would provide two templates for the synthesis of new complementary strands, but exactly how new DNA molecules were constructed was still unclear. In one model, semiconservative replication, the two strands of the double helix separate during DNA replication, and each strand serves as a template from which the new complementary strand is copied. After replication in this model, each double-stranded DNA includes one parental or “old” strand and one daughter or “new” strand. There were two competing models also suggested: conservative and dispersive, which are shown in Figure 9.1.

Figure 9.1 Three Models of DNA replication. In the conservative model, parental DNA strands (blue) remained associated in one DNA molecule while new daughter strands (red) remained associated in newly formed DNA molecules. In the semiconservative model, parental strands separated and directed the synthesis of a daughter strand, with each resulting DNA molecule being a hybrid of a parental strand and a daughter strand. In the dispersive model, all resulting DNA strands have regions of double-stranded parental DNA and regions of double-stranded daughter DNA.
Figure by Parker, N., et.al. (2019) Openstax
Matthew Meselson and Franklin Stahl devised an experiment in 1958 to test which of these models correctly represents DNA replication (Figure 9.2). They grew the bacterium, Escherichia coli for several generations in a medium containing a “heavy” isotope of nitrogen (15N) that was incorporated into nitrogenous bases and, eventually, into the DNA. This labeled the parental DNA. The E. coli culture was then shifted into a medium containing 14N and allowed to grow for one generation. The cells were harvested and the DNA was isolated. The DNA was separated by ultracentrifugation, during which the DNA formed bands according to its density. DNA grown in 15N would be expected to form a band at a higher density position than that grown in 14N. Meselson and Stahl noted that after one generation of growth in 14N, the single band observed was intermediate in position in between DNA of cells grown exclusively in 15N or 14N. This suggested either a semiconservative or dispersive mode of replication. Some cells were allowed to grow for one more generation in 14N and spun again. The DNA harvested from cells grown for two generations in 14N formed two bands: one DNA band was at the intermediate position between 15N and 14N, and the other corresponded to the band of 14N DNA. These results could only be explained if DNA replicates in a semiconservative manner. Therefore, the other two models were ruled out. As a result of this experiment, we now know that during DNA replication, each of the two strands that make up the double helix serves as a template from which new strands are copied. The new strand will be complementary to the parental or “old” strand. The resulting DNA molecules have the same sequence and are divided equally into the two daughter cells.
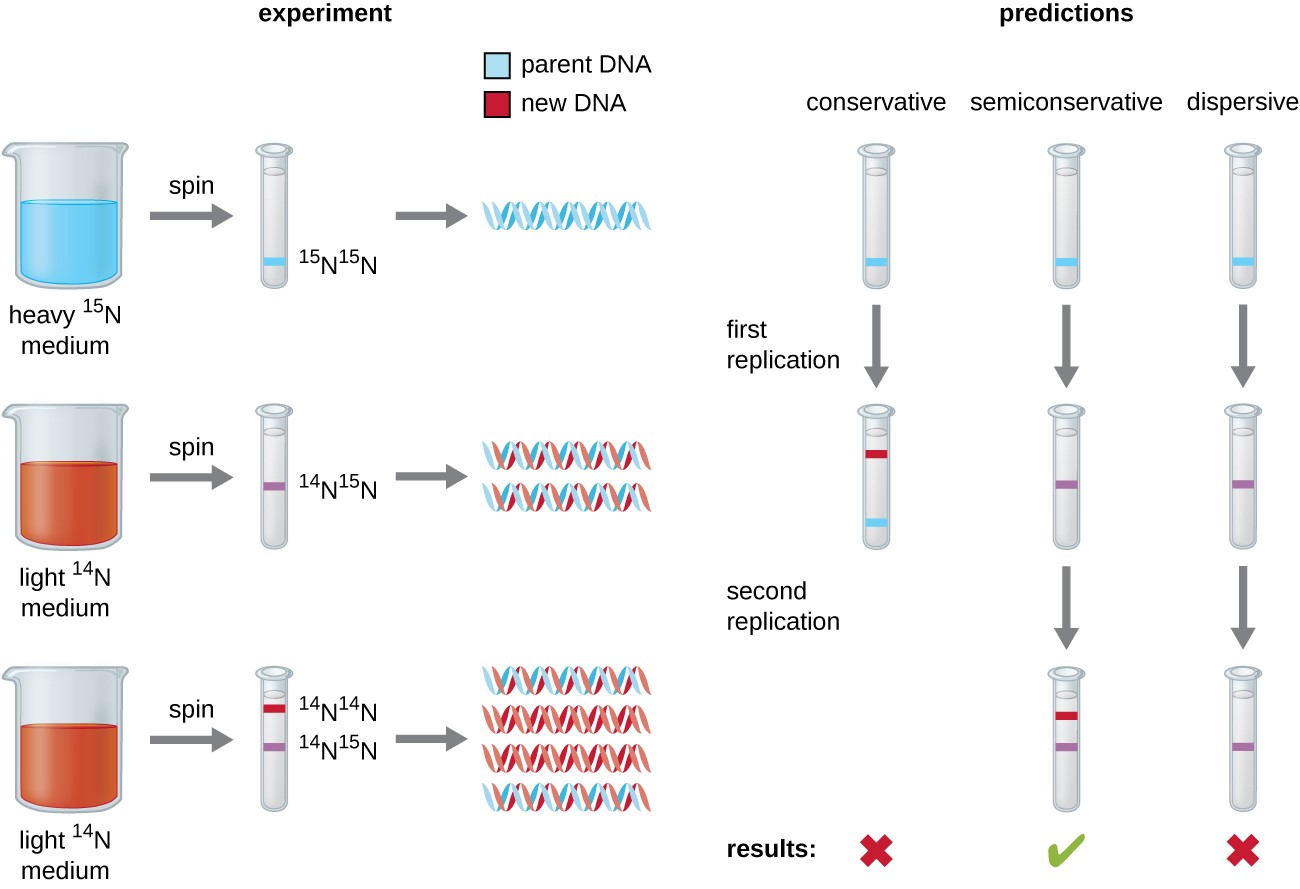
Figure 9.2 Meselson and Stahl experimented with E. coli grown first in heavy nitrogen (15N) then in 14N. DNA grown in 15N (blue band) was heavier than DNA grown in 14N (red band), and sedimented to a lower level on ultracentrifugation. After one round of replication, the DNA sedimented halfway between the 15N and 14N levels (purple band), ruling out the conservative model of replication. After a second round of replication, the dispersive model of replication was ruled out. These data supported the semiconservative replication model.
Figure by Parker, N., et.al. (2019) Openstax
Think about It
- What would have been the conclusion of Meselson and Stahl’s experiment if, after the first generation, they had found two bands of DNA?
Back to the Top
9.2 DNA Replication in Prokaryotes
DNA replication has been well studied in bacteria primarily because of the small size of the genome and the mutants that are available. E. coli has 4.6 million base pairs (Mbp) in a single circular chromosome and all of it is replicated in approximately 42 minutes, starting from a single origin of replication and proceeding around the circle bidirectionally (i.e., in both directions) (Figure 9.3). This means that approximately 1000 nucleotides are added per second. The process is quite rapid and occurs with few errors. E. coli has a single origin of replication, called oriC, on its one chromosome. The origin of replication is approximately 245 base pairs long and is rich in adenine-thymine (AT) sequences.


Figure 9.3 Prokaryotic DNA Replication. Replication of DNA in prokaryotes begins at a single origin of replication, shown in the figure to the left, and proceeds in a bidirectional manner around the circular chromosome until replication is complete. The bidirectional nature of replication creates two replication forks that are actively mediating the replication process. The right hand figure shows a dynamic model of this process. The red and blue dots represent the incorporation of daughter strand nucleotides during the process of replication.
Figures from: Daniel Yuen at David Tribe Derivatives and Catherinea228
Replication Overview
The open regions of DNA that are actively undergoing replication are called replication forks. All the proteins involved in DNA replication aggregate at the replication forks to form a replication complex called a replisome (Table 9.1 and Figure 9.4). DNA replication in the model organism E. coli has been extensively studied, providing a foundation for understanding the diverse mechanisms of genome duplication employed by all organisms. In E. coli, DNA replication is initiated at oriC (Figure 9.3). oriC is ‘melted’ by the action of the DnaA initiator protein to expose two template ssDNA strands that act as platforms for loading the replicative DnaB helicase. One full DnaB hexamer is loaded onto each ssDNA strand with the aid of the helicase loader, DnaC. Additional exposed ssDNA is quickly coated by the ssDNA-binding protein (SSB), which protects DNA and blocks additional DnaB helicase loading. Each DnaB hexamer recruits primase (DnaG), which synthesizes RNA primers used to initiate DNA synthesis, along with the subunits that comprise the replicative DNA polymerase III holoenzyme (PolIII HE). These proteins form the core replisomes that copy the E. coli genome. Once assembled, replisomes replicate bi-directionally away from oriC until, ideally, they undergo programmed disassembly at the termination region, where they encounter ter sites bound by Tus proteins that create ‘replication fork traps’. After completion of DNA replication, the newly synthesized genomes are separated and segregated to daughter cells.
Table 9.1 Enzymes involved in DNA Replication in the prokaryote, E. coli
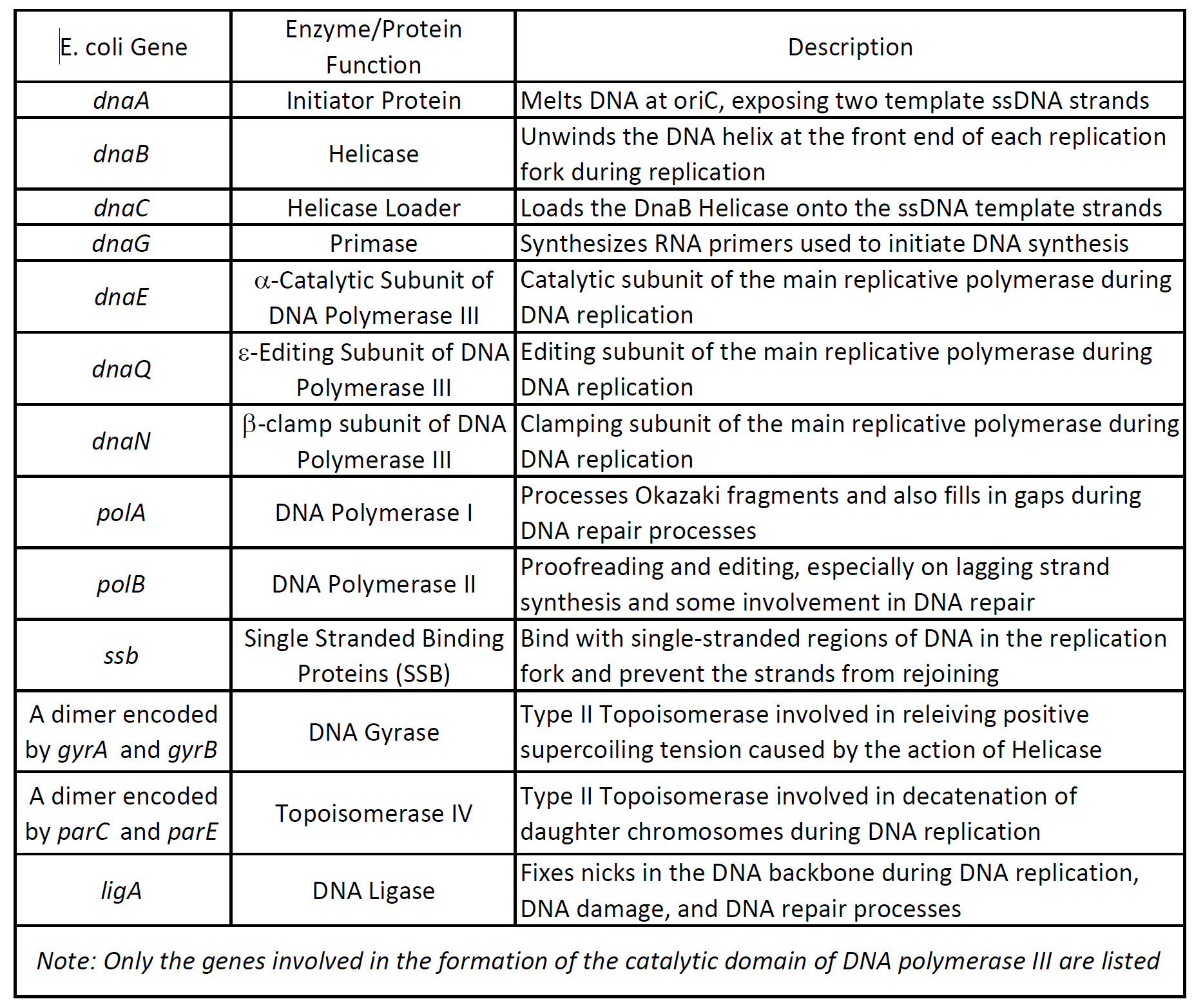

Figure 9.4 General Overview of a DNA Replication Fork. At the origin of replication, topoisomerase II relaxes the supercoiled chromosome. Two replication forks are formed by the opening of the double-stranded DNA at the origin, and helicase separates the DNA strands, which are coated by single-stranded binding proteins to keep the strands separated. DNA replication occurs in both directions. An RNA primer complementary to the parental strand is synthesized by RNA primase and is elongated by DNA polymerase III through the addition of nucleotides to the 3′-OH end. On the leading strand, DNA is synthesized continuously, whereas on the lagging strand, DNA is synthesized in short stretches called Okazaki fragments. RNA primers within the lagging strand are removed by the exonuclease activity of DNA polymerase I, and the Okazaki fragments are joined by DNA ligase.
Figure by Parker, N., et.al. (2019) Openstax
Back to the Top
Primosome Assembly
As noted above, the replication of the bacterial chromosome is initiated at oriC where the initiator protein, DnaA, binds to start the assembly of the enzymatic replisome machine. The early stages of this process involve the assembly of a primosome, that functions to unwind the two strands of DNA at the replication forks and add RNA primers to the DNA templates that will be used by the DNA Polymerase enzymes to begin replication. Subsequent to the remodelling of the replication origin induced by DnaA, the assembly of the bacterial loader-dependent primosome occurs in discrete steps and involves at least four different proteins (initiator protein, helicase, helicase loader protein, and primase) that act in a coordinated and sequential manner (Table 9.1).
The oriC region of prokaryotes contains highly conserved sequence motifs that include an AT-rich box domain that serves as the recognition sequence for the binding of the DnaA initiator protein. Initial binding of DnaA to oriC promotes the melting of the DNA double helix and the recruitment of multiple DnaA subunits that form a helical oligomer along the newly opened single stranded DNA (ssDNA) (Figure 9.5). The DnaA protein contains four major domains. Domains III and IV are integral to binding the ssDNA, while domain I is involved with protein-protein interactions. Domain II forms a flexible linker between the protein interaction domain and the DNA binding domains.
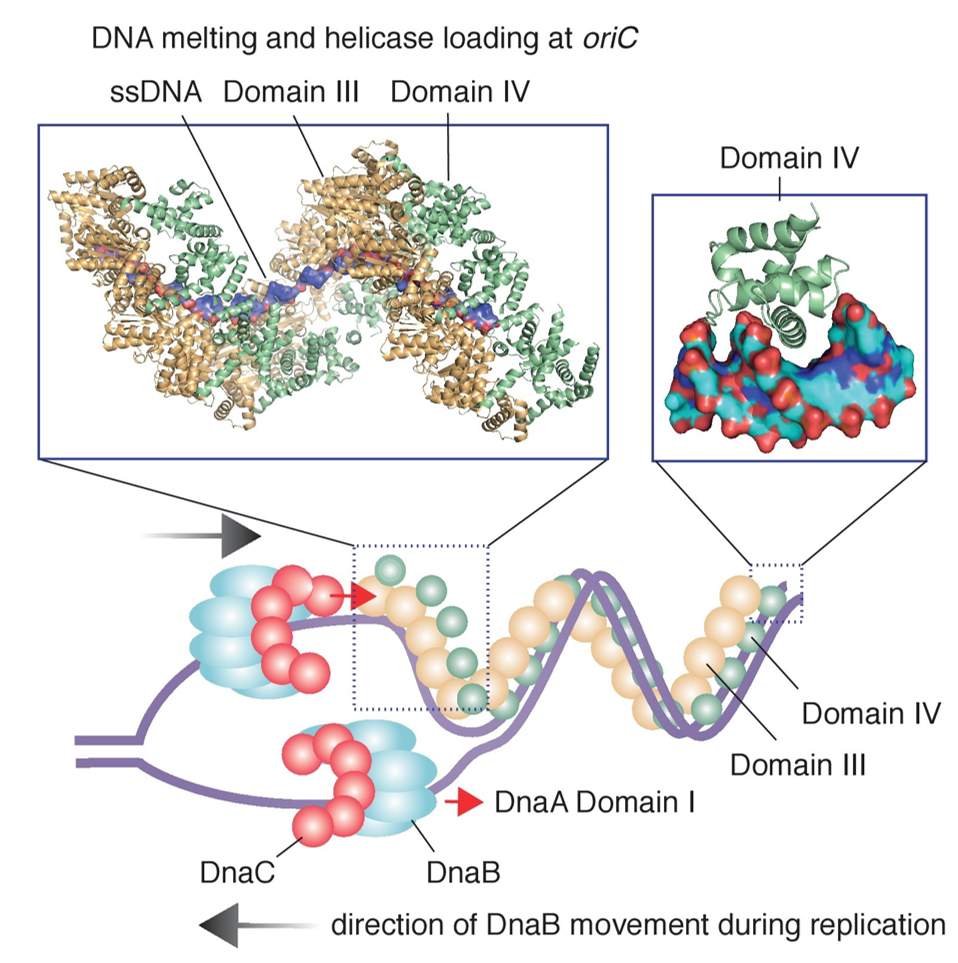
Figure 9.5 Assembly of a Primosome. DNA melting at oriC and loading of the DnaB6–(DnaC)6 helicase–loader complex onto the DNA bubble. Lower schematic: ATP-bound DnaA (the initiator protein) binds to DnaA-boxes via Domain IV, thereby promoting dsDNA to wrap around the DnaA filament, causing torsional strain to the dsDNA. Meantime, Domain III of DnaA binds to one of the two ssDNA strands of DNA unwiding element and stretches the strand. These interactions cause the AT-rich DNA unwinding element to melt, forming a bubble. At the same time, binding of DnaC (the helicase loader) traps DnaB (the helicase) in an open lockwasher conformation, to enable its loading onto ssDNA. DnaC interacts with DnaA at the end of the filament and serves as an adaptor to load one DnaB–DnaC complex. It is not known if closing of DnaB around ssDNA to form a hexameric ring occurs before or concomitantly with dissociation of DnaC. Domain I of DnaA interacts with the N-terminal domain of DnaB, helping to load another DnaB–DnaC on the complementary strand. Upper insets: The helical filament of DnaA formed by Domains III (light orange) and IV (pale green) of Aquifex aeolicus DnaA (PDB: 3R8F) and Domain IV of E. coli DnaA (pale green) bound to dsDNA (PDB: 1J1V). The ssDNA binds in the middle of the DnaA filament via interactions with the AAA+ Domain III of DnaA.
Figure from: Xu, Z-Q. and Dixon, N.E. (2018) Curr Op Struct Biol 53:159-168
In the E.coli system, the helicase loader protein, DnaC, complexed with ATP, binds to hexameric helicase DnaB and forms a DnaB–DnaC complex, which has been confirmed by cryo-electronmicroscope (cryo-EM) studies. The loader protein delivers the helicase on to the melted DNA single strands of the DnaA–oriC nucleoprotein complex at the origin of replication. In vivo, this delivery is associated with the initiator protein, DnaA, whose amino-terminal domain (NTD) is thought to have a role in loading the helicase and helicase loader complex onto the oriC by interacting with helicase, DnaB. After the loader protein dissociates from the helicase ring, the NTD of the helicase interacts with the carboxy-terminal domain (CTD) of the primase and forms a functional primosome. Within the primosome, the helicase (DnaB) acts to unwind the double stranded helix and the primase (DnaG) synthesizes RNA primers on both the leading and lagging DNA strands.
Helicases are motor proteins that move directionally along a nucleic acid phosphodiester backbone, separating two annealed nucleic acid strands such as DNA and RNA, using energy from ATP hydrolysis. There are many helicases, representing the great variety of processes in which strand separation must be catalyzed. Approximately 1% of eukaryotic genes code for helicases. The human genome codes for 95 non-redundant helicases: 64 RNA helicases and 31 DNA helicases. Many cellular processes, such as DNA replication, transcription, translation, recombination, DNA repair, and ribosome biogenesis involve the separation of nucleic acid strands that necessitates the use of helicases. In E. coli, the DnaB Helicase (Figure 9.5) is responsible for unwinding the two parent DNA strands to unwind and separating them from one another to form a “Y”-shaped replication fork. The replication forks are the actual site of DNA copying. During replication within the fork, helix destabilizing proteins, called single stranded binding proteins (SSB), bind to the single-stranded regions preventing the strands from rejoining.
Back to the Top
DNA Polymerases
DNA polymerase enzymes are required for the assembly of the daughter strands along each of the template DNA strands. All DNA polymerases require a DNA template and a primer that is used to begin the replication process. The primer is a short strand of RNA that is placed on the DNA template by the primase enzyme. Recall also that DNA contains two antiparallel strands and that DNA polymerases can only add new nucleotides in the 5′ to 3′ direction when synthesizing the daughter strands of DNA. Since both strands of DNA are replicated simultaneously by the same replisome, the leading strand,where the daughter strand of DNA is moving in the 5′ to 3′ direction, is replicated continuously and flows in the same direction as replisome movement. The lagging strand, that lies in the antiparallel dirction, has to be synthesized in the opposite direction of replisome movement and is created using short bursts of DNA polymerase activity leading to the formation of Okazaki Fragments along the template strand. Thus, the lagging strand must continually be primed with short RNA sequences to maintain the formation of the Okazaki Fragments. The RNA primer sequences must then be replaced by DNA and gaps in the backbone of the DNA must also be repaired.
E. coli has a total of five DNA polymerases. Three of these enzymes are involved in DNA replication (DNA polymerases I, II, and III). DNA polymerase III is the main polymerase involved in both leading strand biosynthesis and the synthesis of the Okazaki Fragments during DNA replication. The DNA polymerase III holoenzyme is comprised of 10 different proteins organized into three functionally distinct, but physically interconnected assemblies: (1) the αεθ core, (2) the β2 sliding clamp, and (3) the δτnγ3-nδ’ΨX clamp loader complex (Figure 9.6). In the polymerase core, α is the polymerase subunit, ε the 3’–5’ proofreading exonuclease and θ is a small subunit that stabilizes ε. After a RNA primer is made by DnaG, the β2clamp is loaded onto the primer terminus by the clamp loader. The α and ε subunits separately bind the clamp, each via a short linear clamp-binding motif (CBM) to the two symmetrically related CBM-binding pockets of β2. Tethered to the clamp, Pol III is able to synthesize DNA at high speed (∼1000 Nt/s) and with much higher processivity(>150 kb).

Figure 9.6 The DNA Replisome. (a) The standard textbook model of a DNA Replisome showing the coupled and highly coordinated processes of leading strand and lagging strand synthesis. DNA polymerase III is connected to the DnaB helicase through the τ subunit of the clamp-loader comples and two or three polymerase cores replicate DNA from both leading strand and lagging strand DNA templates concurrently. The ssDNA in the lagging strand loop is bound by ssDNA binding proteins (SSB). (b) Recent studies have shown that E. coli DNA polymerase III is readily exchangeable at the fork and that leading strand and lagging strand synthesis may not be tightly coupled, or may even be accomplished by different DNA polymerase III holoenzymes. The DnaB helicase can also be decoupled from the DNA polymerase complex and translocate ahead of the apex of the fork.
Figure from: Xu, Z-Q. and Dixon, N.E. (2018) Curr Op Struct Biol 53:159-168
Bacterial replisomes are highly flexible and mobile machines, their dynamics being mediated and controlled by a network of protein–protein interactions of different strengths. Many of the replication proteins are either conformationally flexible or contain flexible or unstructured regions, making structural studies by X-ray crystallography or NMR difficult. However, through decades of efforts, structures of all E. coli replication proteins or their bacterial homologs have been solved as complexes, whole proteins or domains. Recent breakthroughs in single-particle cryo-electron microscopy (cryo-EM) have seen structures determined of large replisome subassemblies, even the whole bacteriophage T7 replisome, though so far only at modest resolution.
Cryo-EM structures of the E. coli Pol III core–clamp–τC (C-terminal domain of the τ subunit of the clamp-loader) complexes on primer–template DNA in both polymerization and proofreading modes were recently solved at 8 and 6.7 Å, respectively, along with structures of a DNA-free complex (Figure 9.7). These structures resemble previously proposed structural models, with some surprises. For example, in the DNA-bound polymerization complex, the β2 clamp becomes almost perpendicular to the DNA strands (Figure 9.7a,b), in contrast to its tilted configuration in the crystal structure of DNA-bound β2. While the Pol III α polymerase subunit binds to DNA in a conformation similar to the crystal structure of DNA-bound Thermus aquaticus (Taq) α, the locations of C-terminal domains (αCTD, comprising the oligonucleotide binding domain, OB, and the τ-binding, TBD, domains) are different. In the Taq α structure and the DNA-free complex, the αCTD is close to the polymerase active site with the OB domain positioned to bind and deliver the ssDNA template into the active site (Figure 9.7c,d). In the DNA-bound cryo-EM structures, these domains are shifted toward the little finger domain of α, the domain that directly contacts the β2 clamp; they are therefore far away from the template strand entering the active site (Figure 9.7e). The OB domain contacts both the little finger and thumb domains of α as well as the β2 clamp and ε. The face of the OB domain that had been thought to be involved in ssDNA template binding now directly faces and is relatively close to the dsDNA. Additionally, ε wedges between the α thumb domain and the clamp. This previously unappreciated interaction network apparently stabilizes the whole complex.

Figure 9.7 Structures of the E. coli polymerase–clamp-τC–DNA complexes. (a) Surface representations of the polymerization (left) and proofreading (right) complexes. The N-terminal domains of α (αNTD, residues 1–963, are colored in deep salmon), and the OB (964–1072) and τ-binding domains (TBD, 1173–1160) of αCTD in brown and dark salmon, respectively, ε in yellow, β2 in aquamarine, θ in orange and τC in slate. The polymerization complex does not include θ, and τC and the αCTD are missing from the proofreading complex. (b) Cartoon representations of complexes showing the differences in the primer–template DNA. In the polymerization complex (left), the DNA has B-form structure, while in the proofreading complex, the primer DNA is frayed with the end of the newly synthesized strand in the active center of ε. The proofreading complex is rotated slightly to show DNA in the active center of ε and the θ subunit is omitted for clarity. (c) Surface representation of αNTD from the DNA-bound polymerization complex (PDB: 5FKV), showing the thumb, palm, fingers, and PHP domains. (d) Positioning of the αCTD in the DNA-free complexes (PDB: 5FKU). (e) Positioning of the αCTD in the DNA-bound polymerization complex (PDB: 5FKV). While the OB domain in the DNA-free complex is close to the active site of Pol III α, it is far away in the DNA-bound complex. The OB domain is colored in marine and the TBD in magenta. The αNTD (gray) in the two complexes shows relatively minor changes compared to αCTD.
Figure from: Xu, Z-Q. and Dixon, N.E. (2018) Curr Op Struct Biol 53:159-168
Overall, there is significant conformational changes in the DNA Polymerase III complex upon binding to the DNA that cause the tail of the polymerase to move from interacting with the clamp in the DNA-bound state, to a position 35 Å away from the clamp in the DNA-free state (Video 9.1). It has been hypothesized that this large conformational change may help the polymerase act as a switch to facilitate the lagging strand synthesis. On the lagging strand, the polymerase repositions to a newly primed site every ∼1000 bp. To do so, the polymerase needs to release both clamp and DNA. The switch-like movement of the polymerase tail may play a part in the release and consequent repositioning of the polymerase at the end of the Okazaki fragment.
Video 9.1 DNA Binding Induces Large Conformational Changes in the DNA Polymerase III Complex. The video shows the linear morphing of the DNA-free to the DNA-bound state showing the large conformation change between the two states. The green subunit is the β-clamp, The α-subunit is shown in orange with the active-site residues in magenta, the α-C-terminal domain (α-CTD shown in brown, the ε-subunit in yellow, and the τ-tail shown in blue.
Video from: Fernandez-Liero, R., et al. (2015) eLife 4:e11134
The proofreading complex is fairly similar to the polymerization complex, with small movements of individual protein components (Figure 9.7a,b). The most significant movements include a rotation and a tilt of duplex DNA against the plane of β2, locking the DNA against the inner surface of the β2 ring (Figure 9.7b). The polymerase thumb domain and ε also move towards the DNA. The thumb domain wedges between two DNA strands with unmatched base pairs, resulting in a highly distorted and frayed DNA substrate. The newly synthesized strand is therefore able to reach the nuclease active site of ε for editing. Considering that the proofreading complex is fairly similar to the polymerization complexes and duplex DNA with two unmatched base-pairs tends to fray, it is proposed that ε works passively by waiting for DNA to reach its nuclease active center when a wrong nucleotide is incorporated rather than responding actively to the misincorporation event. In a complementary single-molecule biophysical study, the clamp-bound Pol III core has been shown to be remarkably stable and processive in the proofreading mode in the absence of incoming dNTPs.
Bacterial replisomes have long been believed to be highly coordinated, highly processive machines capable of copying the whole chromosome without dissociation. Two or three polymerase cores of the same E. coli Pol III HE were believed to synthesize both DNA strands, with the lagging strand polymerase repeatedly being recycled for new Okazaki Fragment synthesis, as described above. Lagging-strand polymerase recycling has been debated to be triggered by various collision or signaling mechanisms in a well-controlled manner that likely involves the movement of the τ-tail region. However, recent studies find that bacterial polymerases also readily exchange at replication forks and that leading-strand and lagging-strand DNA synthesis may not always be tightly coupled. Figure 9.6b depicts a proposed model for this exchange.
Overall, DNA polymerase III is a highly processive enzyme incorporating 600 to 1,000 bases per second with greater that 100,000 bases incorporated per binding event, and an error rate of approximately 1 per million.
DNA polymerase I, aids in the process of lagging strand synthesis, in that, this polymerase removes the RNA primers and incorporates DNA in its place. DNA polymerase II, although not well understood, is thought to play an editing role following lagging strand synthesis by DNA polymerase I. DNA polymerases I and II also play a role in DNA repair, as do DNA polymerases IV and V.
DNA polymerase Iis similar to DNA polymerase III in that it it has 5′ to 3′ polymerase activity and also has 3′ to 5′ exonuclease activity to mediate both the processivity and the DNA proofreading function of the enzyme. In addition, DNA polymerase I also contains a large protein domain called the Klenow fragment that exhibits 5′ to 3′ exonuclease activity (Figure 9.8). The 5′ to 3′ exonuclease activity is responsible for removal of the RNA primers along the lagging strand. The processivity of this enzyme enables the polymerase to refill these gaps with DNA. However, DNA polymerase I is unable to connect the backbone of the newly synthesized Okazaki Fragments with the downstream fragment. The repair of gaps in the DNA backbone is mediated by a DNA ligase enzyme.

Figure 9.8 Structure of E. coli DNA Polymerase I. DNA polymerase I exhibits 5′ to 3′ polymerase activity and 3′ to 5′ exonuclease activity mediating the processivity and proof reading activities of the enzyme. DNA polymerase I also contains a 5′ to 3′ exonuclease activity housed in a special domain of the enzyme called the Klenow fragment. This domain is responsible for removing the RNA primer sequences from newly synthesized DNA. The 5′ to 3′ polymerase activity is then used to replace the RNA primer with DNA. The 3′ to 5′ exonuclease activity ensures that the correct bases are incorporated.
Figure from: Goodsell, D.S. (2015) RCSD PDB-101 Molecule of the Month
Back to the Top
DNA Ligase enzymes seal the breaks in the backbone of DNA that are caused during DNA replication, DNA damage, or during the DNA repair process. The biochemical activity of DNA ligases results in the sealing of breaks between 5′-phosphate and 3′-hydroxyl termini within a strand of DNA. DNA ligases have been differentiated as being ATP-dependent or NAD+-dependent depending on the co-factor (or co-substrate) that is used during their reaction. Typically, more than one type of DNA ligase is found within an organism. As shown in Figure 9.9, the DNA ligase enzyme is covalently modified by the addition of the AMP-moiety to a Lysine residue on the enzyme. The AMP is derived from the ATP or NADH cofactor. The downstream 5′-phosphate at the site of the DNA nick is able to mediate a nucleophilic attack on the AMP-enzyme complex, causing the AMP to transfer to the 5′-phospahte position of the DNA. The AMP serves as a good leaving group for the nucleophilic attack of the upstream 3′-OH with the 5′-phosphate to seal the DNA backbone, and release the AMP.



Figure 9.9 DNA Ligase Reaction. DNA ligases catalyse the crucial step of joining breaks in duplex DNA during DNA repair, replication and recombination, and require either Adenosine triphosphate (ATP) or Nicotinamide adenine dinucleotide (NAD+) as a cofactor. Shown on the upper left is DNA ligase I repairing chromosomal damage. The three visable protein structures are: The DNA binding domain (DBD) which is bound to the DNA minor groove both upstream and downstream of the damaged area. The OB-fold domain (OBD) unwinds the DNA slightly over a span of six base pairs and is generally involved in nucleic acid binding. The Adenylation domain (AdD) contains enzymatically active residues that join the broken nucleotides together by catalyzing the formation of a phosphodiester bond between a phosphate and hydroxyl group. It is likely that all mammalian DNA ligases (Ligases I, III, and IV) have a similar ring-shaped architecture and are able to recognize DNA in a similar manner. The upper right diagram is a high-resolution structure of E. coli LigA in complex with nicked adenylated DNA from PDB 2OWO, visualized by UCSF Chimera. The various domains are indicated by different colours and relate to Pfam domains indicated. The lower diagram depicts the catalytic mechanism of DNA Ligase I. The ATP cofactor forms a covalent linkage with a lysine residue at the α-phosphate position casing the release of diphosphate. The AMP is used to activate the 5′ phosphate group enabling the upstream 3′-OH group to mediate attack on the central phosphorus atom. AMP serves as the leaving group.
Figure on the upper left by: Ellenberger, T at Washington University School of Medicine, St. Louis, MO,Figure on the right by: Pergolizzi, G., Wagner, G.K, and Bowater, R.P. (2016) Biosci Rep 36(5) e00391, and the lower figure by: Showalter, A. (2002)
A summary of the process of DNA replication is shown in Video 9.2
Video 9.2 Overview of the DNA Replication Process
Video from: Yourgenome, animated by Polymime Animation Company, Ltd
Back to the Top
Topoisomerase Enzymes
Unwinding of the double-stranded helix at the replication fork generates winding tension in the DNA in the form of positive supercoils upstream of the replication fork. Enzymes called topoisomerases counteract this by introducing negative supercoils into the DNA in order to relieve this stress in the helical molecule during replication. There are four known topoisomerase enzymes found in E. coli that fall into two major classes, Type I Topoisomerases and Type II Topoisomerases (Figure 9.10). Topoisomerase I and III are Type I topoisomerases, whereas DNA gyrase and Topoisomerase IV are Type II topoisomerases.
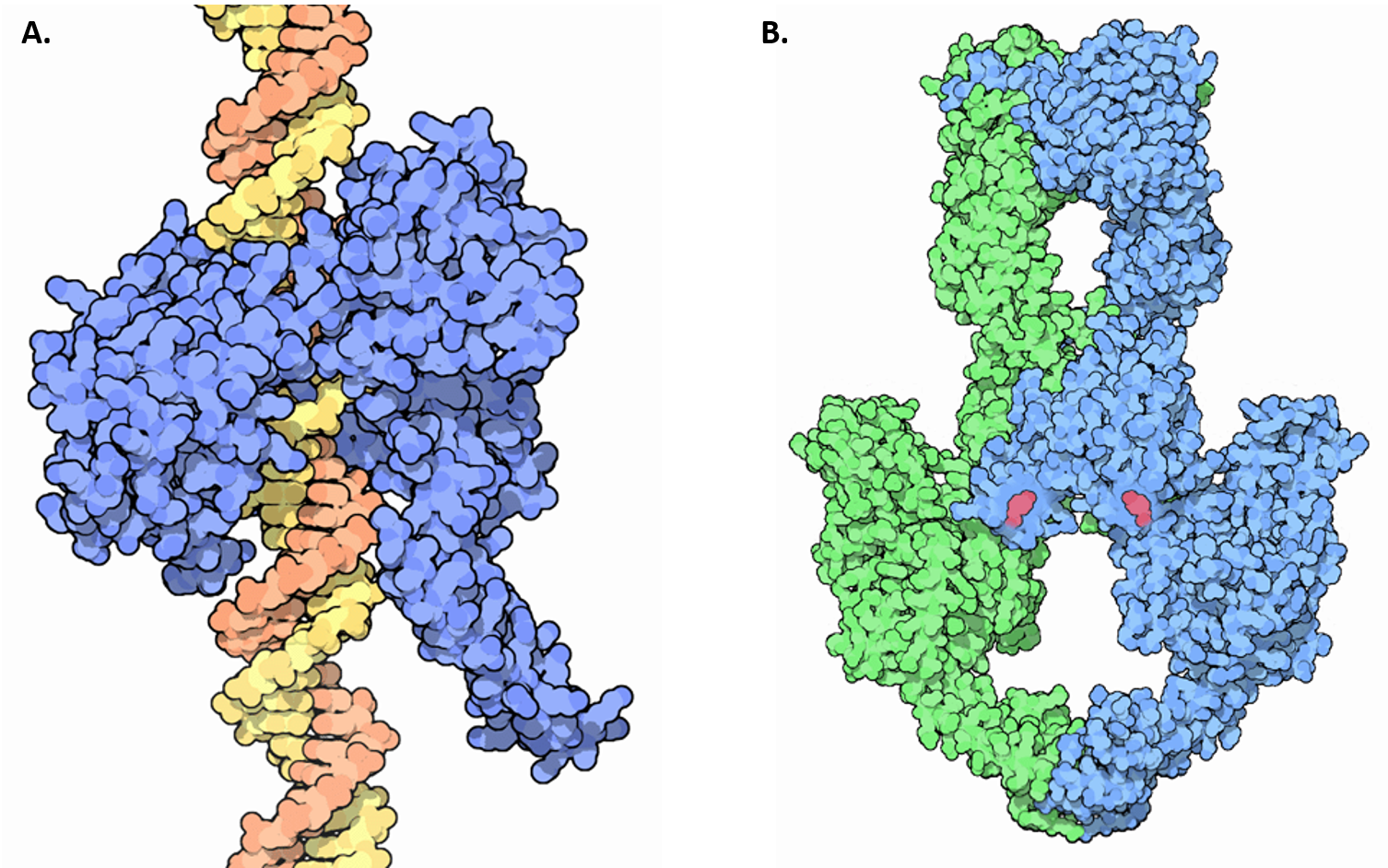
Figure 9.10 Structures of Type I and Type II Topoisomerase. (A) Crystal structure of a Type I Topoisomerase, shown in blue, bound to DNA, shown in orange and yellow. (B) Crystal structure of a Type II Topoisomerase. Topoisomerase II forms a tetrameric structure, shown in green and blue. ATP cofactors (pink) are shown bound to the enzyme.
Goodsell, D.S. (2015) RCSD PDB-101 Molecule of the Month
Type I Topoisomerases relieve tension caused during the winding and unwinding of DNA. One way that they can do this is by making a cut or nick in one strand of the DNA double helix (Figure 9.11). The 5′-phosphoryl side of the nicked DNA strand remains covalently bound to the enzyme at a tyrosine residue, while the 3′-end is held noncovalently by the enzyme. The Type I topoisomerases rotate or spin the 3′-end of the DNA around the intact DNA strand. This releases overwinding in the DNA and effectively releases tension. The enzyme completes the reaction by resealing the phosphodiester backbone or ligating the broken strand back together. Overall, only one strand of the DNA is broken during the reaction mechanism and there is NO requirement of ATP during the reaction. The E. coli Topo I enzyme can only remove negative DNA supercoils, but not positive ones. Thus, this enzyme is not involved in relieving the positive supercoiling caused by the DNA helicase during replication. This is in contrast to eukaryotic Topo I that can relieve both positive and negative supercoiling. Although E. coli Topoisomerase I is not directly involved in relieving the tension caused by DNA replication, it is essential for E. coli viability. It is thought to help balance the actions of the Type II topoisomerases and help maintain optimal supercoiling density within the chromosomal DNA. Thus, Topo I is thought to help maintain homeostatic balance of chromosome supercoiling within E. coli. Topo III, which is also a Type I Topoisomerase, appears to play a role in the decatenation of the daughter chromosomes during DNA replication, but does not play a role in the relaxation of supercoiling.
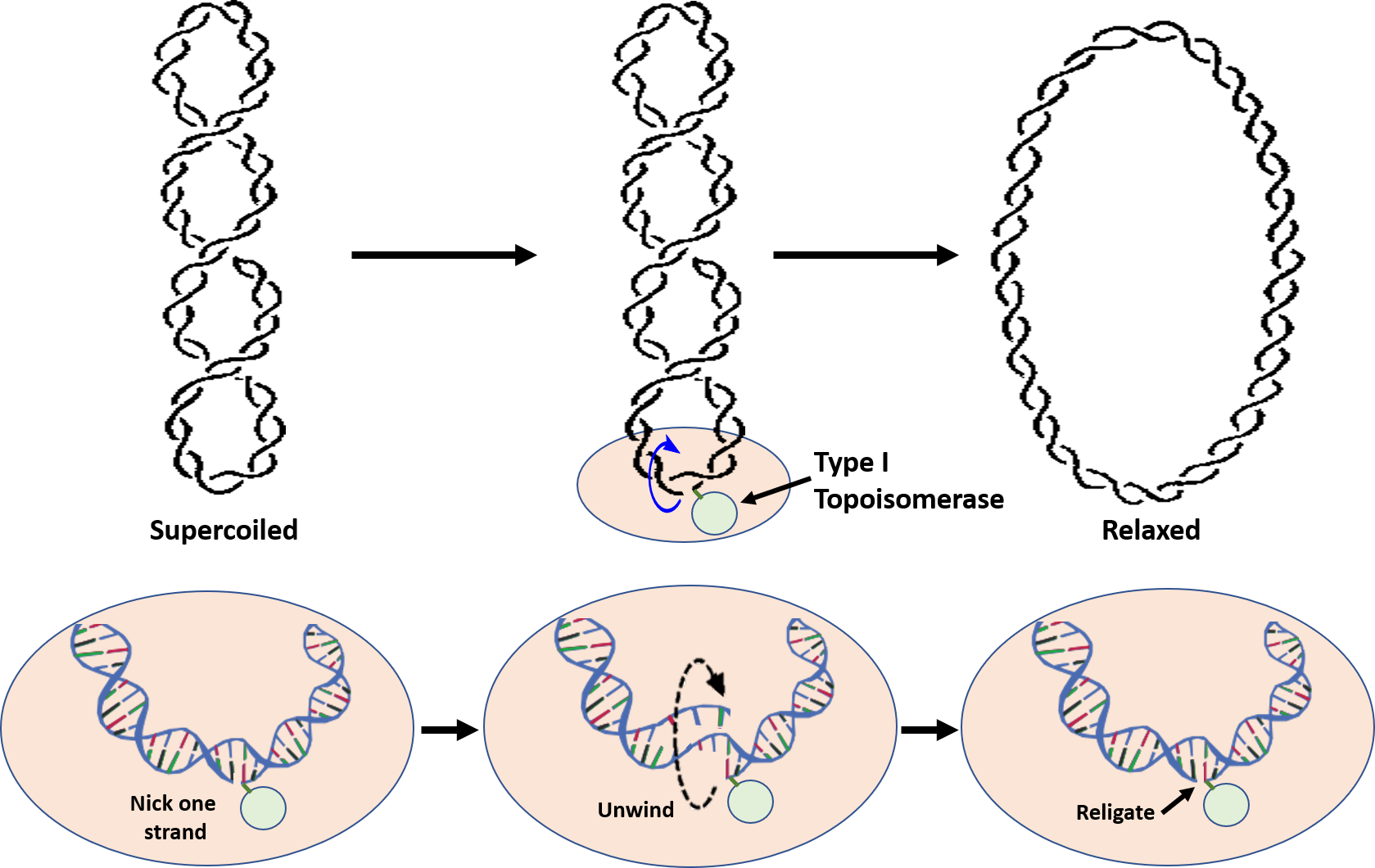
Figure 9.11 Reaction of Type I Topoisomerases. During the reaction, Type I Topoisomerases nick one strand of the DNA. One end of the nicked DNA is covalently attached to the enzyme, shown in light green in the lower diagrams. The other end is held non-covalently and rotated around the double helix to unwind the supercoiling and relax the DNA. Once supercoiling tension is released, the backbone is resealed and the Topoisomerase I enzyme is released.
Figure remixed from: Notahelix and JoKalliauer, andthe National Human Genome Research Institute
Type II Topoisomerases have multiple functions within the cell. They can increase or decrease winding tension within the DNA or they can unknot or decatanate DNA that has become tangled with another strand (Figure 9.12). It does so by a more dangerous method than their Type I counterparts, by breaking both strands of the DNA during their reaction mechanism. The enzyme is covalently attached to both broken sides while the other DNA helix is passed through the break. The double strand break is then resealed.
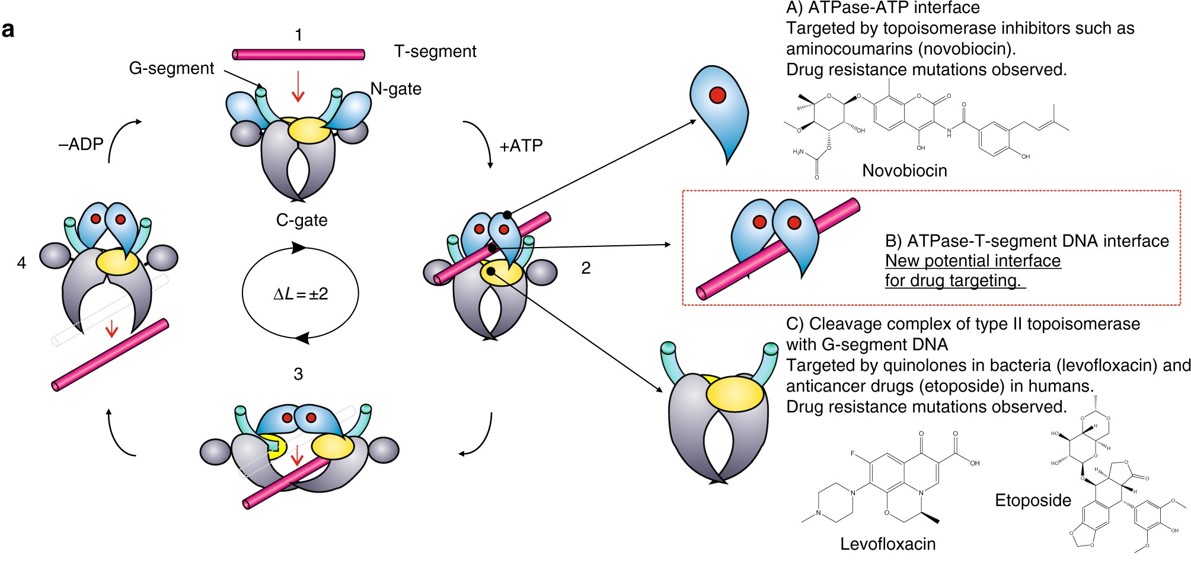
Figure 9.12 Reaction of Type II Topoisomerases. The proposed type II topoisomerase reaction cycle is exemplified by topoisomerase IV. Topoisomerase IV subunits are denoted in grey, cyan, and yellow. The gate or G-DNA is in green and the transported or T-DNA is in mauve. ATP bound to the ATPase domains is denoted by a red dot. In step 1, the G-DNA binds with the enzyme. ATP and the T-DNA segment associate with the enzyme in step 2. In step 3, the G-DNA is cleaved and the T-DNA is passed through the break. Drug-targetable domains within the type II topoisomerase complex are highlighted in subsections A, B and C with examples on the right-hand side of the figure.
Figure modified from: Laponogov, I., et al. (2018) Nature Communications 9:2579.
DNA gyrase, is the type II topoisomerase enzyme that is primarily involved in relieving positive supercoiling tension that results due to the helicase unwinding at the replication fork. Type II Topoisomerases, especially Topo IV, also address a key mechanistic challenge that faces the bacterial replisome during termination of DNA replication. The circular nature of the bacterial chromosome dictates that a pair of replisomes that initiate from a single origin of replication will eventually converge on each other in a head-to-head orientation.Positive supercoiling accumulates between the two replisomes as they converge, but the activity of DNA gyrase, which normally removes positive supercoils, becomes limited by the decreasing amount of template DNA available.Instead, supercoils may diffuse behind the replisomes, forming precatenanes between newlyr eplicated DNA; in E.coli these must be resolved byTopo IV for chromosome segregation to occur.
Tus Proteins and the Termination of Replication
Proper termination of DNA replication is important for genome stability. E. coli replication terminates in the region opposite oriC. There are ten 23-bp termination (Ter) sites in the region with some sequence variations that determine their binding affinities for the monomeric termination protein Tus (Figure 9.13). Tus binds to Ter with high affinity in 1:1 ratio, and Tus–Ter can further form a very stable ‘lock’ complex if cytosine-6 of the strictly conserved G–C(6) base pair of Ter is flipped out of the DNA duplex and bound in a preformed cytosine-binding pocket of Tus (Figure 9.13b). The Tus–Ter lock complex is polar with a permissive face that allows the replisome to pass unhindered and a non-permissive face that can block the replisome. The ten Ter sites are organized as two oppositely orientated groups of five, allowing the replisome to pass the first group and be blocked at the second. This ensures that the two replication forks converge in the terminus region for proper chromosome segregation.

Figure 9.13 Mechanisms of replisome blockage by Tus–Ter replication termination complexes.(a) Schematic representation of the E. coli chromosome, showing positions of oriC and Ter sites. The clockwise moving fork passes through the permissive sites shown in green and is arrested at the non-permissive sites shown in red. (b) Schematic representation of structure of the ‘locked’ Tus-Ter complex (PDB: 2I06), showing cytosine-6 in its binding pocket in Tus. (c) Interactions of residue Arg198 of Tus with both strands of Ter in complexes with double-stranded wild-type Ter (PDB: 2I05, left) and the Tus–Ter UGLC complex (GC(6) base pair inverted; PDB: 4XR3, right).
Figure from: Xu, Z-Q. and Dixon, N.E. (2018) Curr Op Struct Biol 53:159-168
Back to the Top
9.3 DNA Replication of Extrachromosomal Elements: Plasmids and Viruses
To copy their nucleic acids, plasmids and viruses frequently use variations on the pattern of DNA replication described for prokaryote genomes. We will focus here on one style known as the rolling circle method.
Rolling Circle Replication
Whereas many bacterial plasmids replicate by a process similar to that used to copy the bacterial chromosome, other plasmids, several bacteriophages, and some viruses of eukaryotes use rolling circle replication (Figure 9.14). The circular nature of plasmids and the circularization of some viral genomes on infection make this possible. Rolling circle replication begins with the enzymatic nicking of one strand of the double-stranded circular molecule at the double-stranded origin (dso) site. In bacteria, DNA polymerase III binds to the 3′-OH group of the nicked strand and begins to unidirectionally replicate the DNA using the un-nicked strand as a template, displacing the nicked strand as it does so. Completion of DNA replication at the site of the original nick results in full displacement of the nicked strand, which may then recircularize into a single-stranded DNA molecule. RNA primase then synthesizes a primer to initiate DNA replication at the single-stranded origin (sso) site of the single-stranded DNA (ssDNA) molecule, resulting in a double-stranded DNA (dsDNA) molecule identical to the other circular DNA molecule.

Figure 9.14 Rolling Circle Replication. The process of rolling circle replication is initiated by a single stranded nick in the DNA. Within prokaryotes, DNA polymerase III is utilized to generate the daughter strand. DNA ligase rejoins nicks in the backbone and enables the initiation of DNA synthesis of the second daughter strand.
Figure byParker, N., et.al. (2019) Openstax
Back to the Top
9.4 DNA Replication in Eukaryotes
The Cell Cycle
The cell cycle is an ordered series of events involving cell growth and cell division that produces two new daughter cells. Cells on the path to cell division proceed through a series of precisely timed and carefully regulated stages of growth, DNA replication, and division that produce two genetically identical cells. The cell cycle has two major phases: interphase and the mitotic phase (Figure 9.15). During interphase, the cell grows and DNA is replicated. During the mitotic phase, the replicated DNA and cytoplasmic contents are separated and the cell divides. Watch this video about the cell cycle: http://openstax.org/l/biocellcyc

Figure 9.15 Diagram of the Cell Cycle. A cell moves through a series of phases in an orderly manner. During interphase, G1 involves cell growth and protein synthesis, the S phase involves DNA replication and the replication of the centrosome, and G2 involves further growth and protein synthesis. The mitotic phase follows interphase. Mitosis is nuclear division during which duplicated chromosomes are segregated and distributed into daughter nuclei. Usually the cell will divide after mitosis in a process called cytokinesis in which the cytoplasm is divided and two daughter cells are formed.
Figure from Fowler, S., et.al. (2013) Openstax
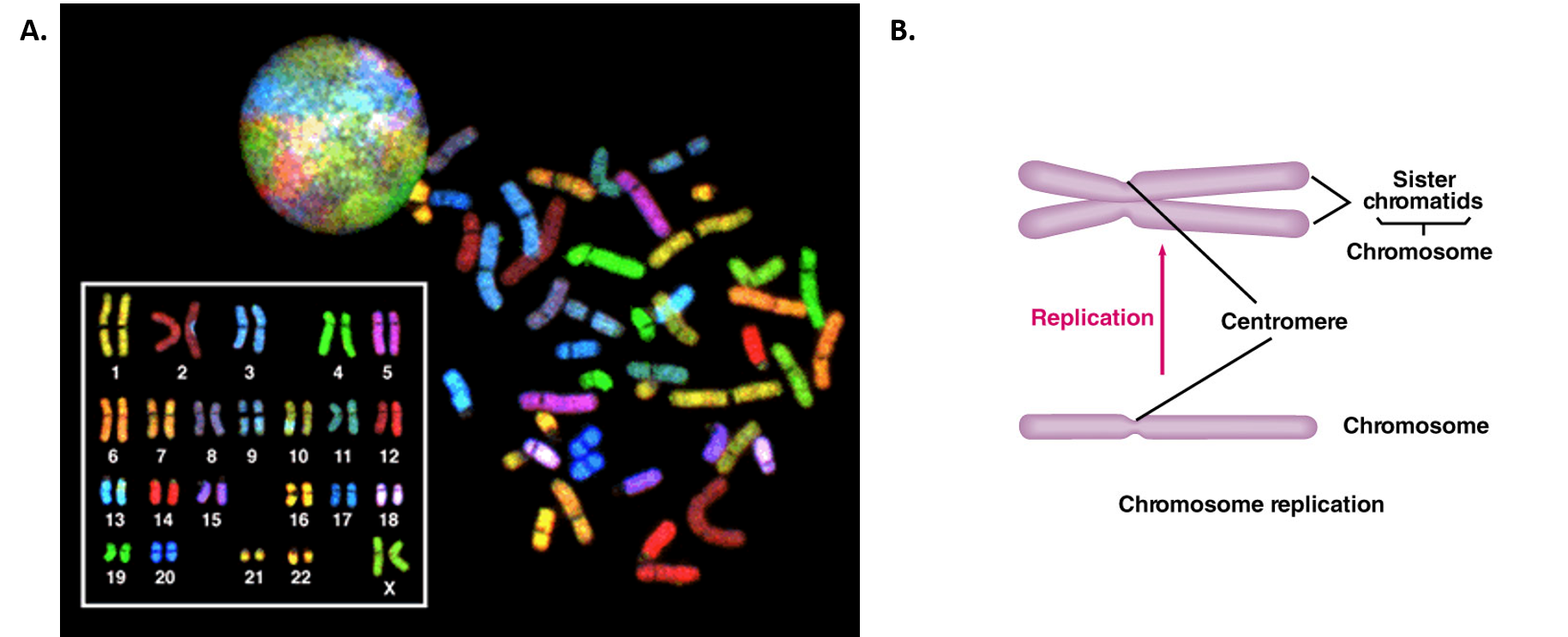
During interphase, the cell undergoes normal processes while also preparing for cell division. For a cell to move from interphase to the mitotic phase, many internal and external conditions must be met. The three stages of interphase are called G1, S, and G2. The first stage of interphase is called the G1 phase, or first gap, because little change is visible. However, during the G1 stage, the cell is quite active at the biochemical level. The cell is accumulating the building blocks of chromosomal DNA and the associated proteins, as well as accumulating enough energy reserves to complete the task of replicating each chromosome in the nucleus. Throughout interphase, nuclear DNA remains in a semi-condensed chromatin configuration. In the S phase (synthesis phase), DNA replication results in the formation of two identical copies of each chromosome—sister chromatids—that are firmly attached at the centromere region (Figure 9.16B). At this stage, each chromosome is made of two sister chromatids and is a duplicated chromosome. The centrosome is duplicated during the S phase. The two centrosomes will give rise to the mitotic spindle, the apparatus that orchestrates the movement of chromosomes during mitosis. In mammals, the centrosome consists of a pair of rod-like centrioles at right angles to each other. Centrioles help organize cell division. Centrioles are not present in the centrosomes of many eukaryotic species, such as plants and most fungi.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 9.16 Human Chromosome Structure. (A) Shows a spectral karyogram of a normal human female. Humans have a total of 23 pairs of chromosomes for a total of 46. Each pair of chromosomes are referred to as homologous chromosomes as they contain copies of the same gene regions. Each of the homologous pairs of chromosomes are stained the same color. Chromosomes are shown in their condensed, unreplicated state. (B) Shows a schematic diagram of a single chromosome before (lower diagram) and after (upper diagram) replication. Upon replication, the identical copies of the chromosome are called sister chromatids and are linked together at the centromere structure.
Figure A from: The National Human Genome Research Institute, and Figure B from: The School of Biomedical Sciences Wiki
{kind=link}
{kind=link}
In the G2 phase, or second gap, the cell replenishes its energy stores and synthesizes the proteins necessary for chromosome manipulation. Some cell organelles are duplicated, and the cytoskeleton is dismantled to provide resources for the mitotic spindle. There may be additional cell growth during G2. The final preparations for the mitotic phase must be completed before the cell is able to enter the first stage of mitosis. To make two daughter cells, the contents of the nucleus and the cytoplasm must be divided. The mitotic phase is a multistep process during which the duplicated chromosomes are aligned, separated, and moved to opposite poles of the cell, and then the cell is divided into two new identical daughter cells. The first portion of the mitotic phase, mitosis, is composed of five stages, which accomplish nuclear division. The second portion of the mitotic phase, called cytokinesis, is the physical separation of the cytoplasmic components into two daughter cells.
If cells are not traversing through one of the phases of interphase or mitosis, they are said to be in G0 or a resting state. If cells enter G0 permanently, they are said to have entered a stage of replicative senescence and will no longer be maintained for long term viability within the organism.
Progression of cells through the cell cycle requires the coordinated actions of specific protein kinases, known as cyclin-dependent kinases. Cyclin-dependent kinases are usually abbreviated as CDK or CDC proteins. CDK/CDC proteins require the binding of a regulatory cyclin protein to become activated (Figure 9.17). The major cyclin proteins that drive the cell cycle in the forward direction, are expressed only at discrete times during the cell cycle. When activated by a cyclin counterpart, CDK/CDC enzymes phosphorylate downstream targets involved with cell cycle progression. For example, the primary cyclin-CDK complex involved in the initiation of DNA replication during S-phase is the CyclinE-CDK2 complex. CDK2 is activated by the expression and binding of Cyclin E during late G1 phase. This causes CDK2 to phosphorylate downstream targets, including the retinoblastoma tumor suppressor protein, pRb. pRB normally binds and inhibits the the activity of transcription factors from the E2F family. Following the release of E2F transcription factors from pRb, E2Fs activate the transcription of genes involved in DNA replication and the leads to the progression of cells into S-phase.
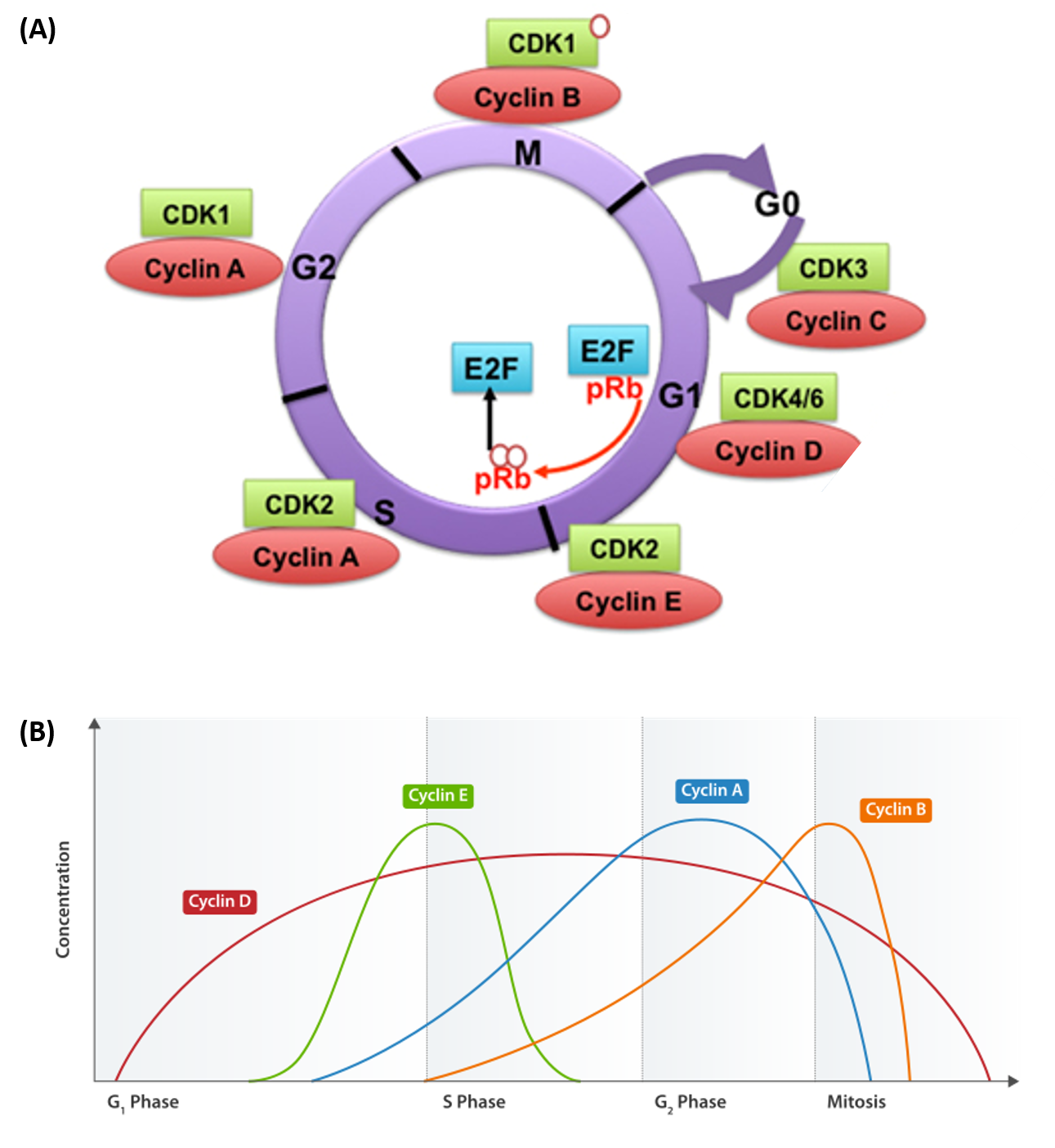
Figure 9.17 Cyclin Dependent Kinases (CDKs) and their Cyclin Regulatory Subunits. (A) CDK-cyclin complexes with direct functions in regulating the cell cycle are shown. CDK3/cyclin C drives cell cycle entry from G0. CDK4/6/cyclin D complexes initiate phosphorylation of the retinoblastoma protein (pRb) and promote the activation of CDK2/cyclin E complex. In late G1, CDK2/cyclin E complex completes phosphorylation and inactivation of pRb, which releases the E2F transcription factors and G1/S transition takes place. DNA replication takes place in S phase. CDK2/cyclin A complex regulates progression through S phase and CDK1/cyclin A complex through G2 phase in preparation for mitosis (M). Mitosis is initiated by CDK1/cyclin B complex. (B) Shows the cyclical nature of cyclin expression during cell cycle progression. Cyclin abundance is regulated by transcriptional expression and rapid protein degradation. Thus, their biological activity is targeted at very specific time points during cell cyle progression.
Figure A from: Aleem, E., and Arceci, R.J. (2015) Front. Cell and Dev 3 (16) and Figure B from: Cyclinexpression_waehrend-Zellzyklus
{kind=link}
Back to the Top
Replication Initiation
Origin organization, specification, and activation in eukaryotes are more complex than in bacterial or archaeal kingdoms and significantly deviate from the paradigm established for prokaryotic replication initiation. The large genome sizes of eukaryotic cells, which range from 12 Mbp in S. cerevisiae to 3 Gbp in humans, necessitates that DNA replication starts at several hundred (in budding yeast) to tens of thousands (in humans) origins to complete DNA replication of all chromosomes during each cell cycle (Figure 9.18).


Figure 9.18. Eukaryotic chromosomes are typically linear, and each contains multiple origins of replication. Lefthand figure is a graphic representation of eukayotic origins of replication, while the righthand image is a Cryo-electron micrograph image
The figure on the lefthand side is from: Parker, N. et al. and the figure on the righthand side is from: Fritensky, B. and Brien, N
With the exception of S.cerevisiae and related Saccharomycotina species, eukaryotic origins do not contain consensus DNA sequence elements but their location is influenced by contextual cues such as local DNA topology, DNA structural features, and chromatin environment. Nonetheless, eukaryotic origin function still relies on a conserved initiator protein complex to load replicative helicases onto DNA during the late M and G1 phases of the cell cycle, a step known as origin licensing. In contrast to their bacterial counterparts, replicative helicases in eukaryotes are loaded onto origin duplex DNA in an inactive, double-hexameric form and only a subset of them (10–20% in mammalian cells) is activated during any given S phase, events that are referred to as origin firing. The location of active eukaryotic origins is therefore determined on at least two different levels, origin licensing to mark all potential origins, and origin firing to select a subset that permits assembly of the replication machinery and initiation of DNA synthesis. The extra licensed origins serve as backup and are activated only upon slowing or stalling of nearby replication forks, ensuring that DNA replication can be completed when cells encounter replication stress. Together, the excess of licensed origins and the tight cell cycle control of origin licensing and firing embody two important strategies to prevent under- and overreplication and to maintain the integrity of eukaryotic genomes.
Eukaryotic DNA Polymerase Enzymes
Similar to DNA replication in prokaryotes, DNA replication in eukaryotes occurs in opposite directions between the two new strands at the replication fork. Within eukaryotes, two replicative polymerases synthesize DNA in opposing orientations (Figure 9.16). Polymerase ε (epsilon)synthesizes DNA in a continuous fashion, as it is “pointed” in the same direction as DNA unwinding. Similar to bacterial replication, this strand is known as the leading strand. In contrast, polymerase δ (delta) synthesizes DNA on the opposite template strand in a fragmented, or discontinuous, manner and this strand is termed the lagging strand. The discontinuous stretches of DNA replication products on the lagging strand are known as Okazaki fragments and are about 100 to 200 bases in length at eukaryotic replication forks. Owing to the “lagging” nature, the lagging strand generally contains a longer stretch of ssDNA that is coated by single-stranded binding proteins, which stabilizes ssDNA templates by preventing secondary structure formation or other transactions at the exposed ssDNA. In eukaryotes, ssDNA stabilization is maintained by the heterotrimeric complex known as replication protein A (RPA) (Figure 9.19). Each Okazaki fragment is preceded by an RNA primer, which is displaced by the procession of the next Okazaki fragment during synthesis. In eukaryotic cells, a small amount of the DNA segment immediately upstream of the RNA primer is also displaced, creating a flap structure. This flap is then cleaved by endonucleases (such as Fen1, discussed later). At the replication fork, the gap in DNA after removal of the flap is sealed by DNA ligase I. Owing to the relatively short nature of the eukaryotic Okazaki fragment, DNA replication synthesis occurring discontinuously on the lagging strand is less efficient and more time consuming than leading-strand synthesis.

Figure 9.19 The Eukaryotic Replisome Complex Coordinates DNA Replication. Replication on the leading and lagging strands is performed by Pol ε and Pol δ, respectively. Many replisome factors (including the FPC [fork protection complex], Claspin, And1, and RFC [the replication factor C clamp loader]) are charged with regulating polymerase functions and coordinating DNA synthesis with unwinding of the template strand by Cdc45-MCM [mini-chromosome maintenance]-GINS [go-ichi-ni-san]. The replisome also associates with checkpoint proteins as DNA replication and genome integrity surveillance mechanisms.
Figure from: Lemanm A.R. and Noguchi, E. (2013) Genes 4(1):1-32
At the eukaryotic replication fork, three distinct replicative polymerase complexes contribute to canonical DNA replication: α, δ, and ε. These three polymerases are essential for viability of the cell. Because DNA polymerases require a primer on which to begin DNA synthesis, first, polymerase α (Pol α) acts as a replicative primase. Pol α is associated with an RNA primase and this complex accomplishes the priming task by synthesizing a primer that contains a short ~10-nucleotide RNA stretch followed by 10 to 20 DNA bases. Importantly, this priming action occurs at replication initiation at origins to begin leading-strand synthesis and also at the 5′ end of each Okazaki fragment on the lagging strand.
However, Pol α is not able to continue DNA replication. From in vitro studies, it was observed that DNA replication must be “handed off” to another polymerase to continue synthesis. The polymerase switching requires clamp loaders. Initially, it was thought that Pol δ performed leading-strand replication and that Pol α completed each Okazaki fragment on the lagging strand. Using mutator polymerase variants and mapping nucleotide misincorporation events, Kunkel and colleagues found that Pol ε and Pol δ mutations lead to mismatched nucleotide incorporation only on the leading and lagging strands, respectively. Thus, normal DNA replication requires the coordinated actions of three DNA polymerases: Pol α for priming synthesis, Pol ε for leading-strand replication, and Pol δ for generating Okazaki fragments during lagging-strand synthesis (Figure 9.16).
In eukaryotes, DNA polymerases are grouped into seven families (A, B, C, D, X, Y, and RT). Crystal structures of the three nuclear replicative DNA polymerases demonstrate that they belong to the B family (Figure 9.17). All three replicative DNA polymerases are multi-subunit enzymes (Table 9.2)
Table 9.2 Subunits of the Major Eukaryotic Replicative DNA Polymerases
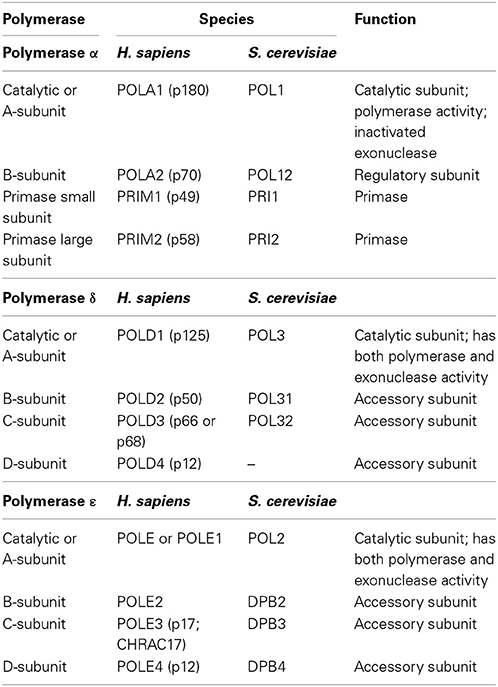
Table from:Doublié, S. and Zahn, K.E. (2014) Front. Microbiol 5:444
Back to the Top
All B family polymerases are composed of five subdomains: the fingers, thumb, and palm which constitute the core of the enzyme, as well as an exonuclease domain and an N-terminal domain (NTD). The palm, a highly conserved fold composed of four antiparallel β strands and two helices, harbors two strictly conserved catalytic aspartates located in motif A, DXXLYPS and motif C, DTDS (Figure 9.20). This fold is shared by a very large group of enzymes, including DNA and RNA polymerases, reverse transcriptases, CRISPR polymerase, and even reverse (3′–5′) transferases. In contrast, the thumb and fingers subdomains exhibit substantially more structural diversity. The fingers undergo a conformational change upon binding DNA and the correct incoming nucleotide. This movement allows residues in the fingers subdomain to come in contact with the nucleotide in the nascent base pair. The thumb holds the DNA duplex during replication and plays a part in processivity. The exonuclease domain carries a 3′–5′ proofreading activity, which removes misincorporated nucleotides. The NTD seems to be devoid of catalytic activity. In pol δ the NTD comprises three motifs: one has a topology resembling an OB fold, a single-stranded DNA binding motif, and another bears an RNA-binding motif (RNA Recognition Motif or RRM). The NTD likely plays a role in polymerase stability and fidelity through its interactions with other domains.
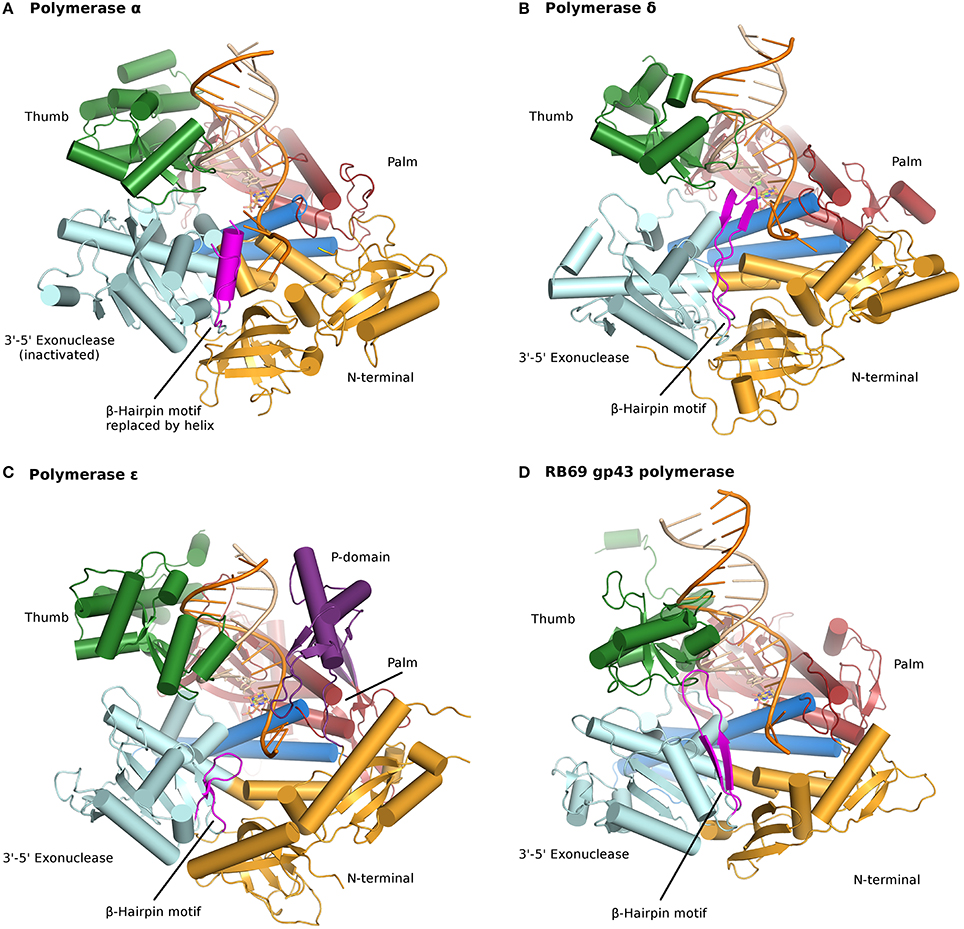
Figure 9.20 Ternary complexes of polymerases α, δ, ε, and RB69 gp43 are illustrated from identical orientations for comparison. The thumb (green) and fingers (dark blue) domains grasp the duplex nucleic acid (primer shown in beige, template in orange) against the palm domain (red). The N-terminal domain appears in gold, adjacent to the 3′–5′ exonuclease domain (cyan). (A) Polymerase α (PDBID 4FYD) binds an RNA/DNA hybrid, where the wide, shallow minor groove of A-form DNA is apparent near the thumb. The 3′–5′ exonuclease domain is devoid of activity. A helical region (magenta) in the inactivated exonuclease domain stabilizes the 5′end of the template. (B) Polymerase δ (PDBID 3IAY) harbors a large β hairpin motif (magenta), which is important in switching the primer strand from the polymerase active site to the exonuclease active site in the event of proofreading. (C) Polymerase ε (PDBID 4M8O) wields a unique P-domain (purple), which endows the polymerase with increased processivity. Interestingly, the β hairpin motif is atrophied in pol ε. (D) Conservation of the family B DNA polymerase fold, and domain organization, is evident when the model enzyme from bacteriophage RB69 gp43 (PDBID 2OZS) is viewed along with the three eukaryotic replicative polymerases. The domain delineation for each polymerase is given in Table S1. Figure was made with PyMOL (The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC.)
Figure from: Doublié, S. and Zahn, K.E. (2014) Front. Microbiol 5:444
DNA polymerases require additional factors to support DNA replication in vivo. DNA polymerases have a semi-closed hand structure, which allows them to load onto DNA and translocate. This structure permits DNA polymerase to hold the single-stranded template, incorporate dNTPs at the active site, and release the newly formed double strand. However, the conformation of DNA polymerases does not allow for their stable interaction with the template DNA. To strengthen the interaction between template and polymerase, DNA sliding clamps have evolved, promoting the processivity of replicative polymerases. In eukaryotes, this sliding clamp is a homotrimer known as proliferating cell nuclear antigen (PCNA), which forms a ring structure. The PCNA ring has polarity with a surface that interacts with DNA polymerases and tethers them securely to DNA. PCNA-dependent stabilization of DNA polymerases has a significant effect on DNA replication because it enhances polymerase processivity up to 1,000-fold (Figure 9.19).
The DNA helicases (MCM proteins) and polymerases must also remain in close contact at the replication fork (Figure 9.19). If unwinding occurs too far in advance of synthesis, large tracts of ssDNA are exposed. This can activate DNA damage signaling or induce aberrant DNA repair processes. To thwart these problems, the eukaryotic replisome contains specialized proteins that are designed to regulate the helicase activity ahead of the replication fork. These proteins also provide docking sites for physical interaction between helicases and polymerases, thereby ensuring that duplex unwinding is coupled with DNA synthesis.
Control of Origin Firing
Origin usage in eukaryotes can be dynamic, with origin firing at different sites depending on cell type and developmental stage. Nevertheless, the mechanism of replisome assembly and origin firing is highly conserved. During late mitosis and G1phase, cell cycle proteins, such as Cdc6, associate with Ori sites throughout the genome and recruit the helicase enzymes, MCMs 2-7 (Figure 9.21A). At this time, double hexamers of the MCM2-7 complex are loaded at replication origins. This generates a pre-replication complex (pre-RC). Origins with an associated pre-RC are considered licensed for replication. Licensed replication origins can then be “fired,” when replication actually initiates at the Ori. Origin firing is brought about by multiple phosphorylation events carried out by the cyclin E-CDK2 complex at the onset of S phase and by other cyclin-dependent kinases (CDKs) prior to individual origin firing (Figure 9.21B). Cyclin-dependent kinases (CDKs) are the families of protein kinases first discovered for their role in regulating the cell cycle. They are also involved in regulating transcription, mRNA processing, and the differentiation of nerve cells. CDKs are activited through the binding of an associated cyclin regulatory protein. Without a cyclin, CDKs exhibit little kinase activity. Following the phosphorylation of the pre-RC, origin melting occurs and DNA unwinding by the helicase generates ssDNA, exposing a template for replication (Figure 9.21C). The replisome then begins to form with the localization of replisome factors such as Cdc45. DNA synthesis begins on the melted template, and the replication machinery translocates away from the origin in a bidirectional manner.

Figure 9.21 MCM2-7 loads onto DNA at replication origins during G1 and unwinds DNA ahead of replicative polymerases. (A) The combined activities of Cdc6 and Cdt1 bring MCM complexes (shown as blue circles of varying shades) to replication origins. (B) CDK/DDK-dependent phosphorylation of pre-RC components leads to replisome assembly and origin firing. Cdc6 and Cdt1 are no longer required and are removed from the nucleus or degraded (C) MCMs and associated proteins (GINS and Cdc45 are shown) unwind DNA to expose template DNA. At this point replisome assembly can be completed and replication initiated. “P” indicates phosphorylation.
Figure from: Lemanm A.R. and Noguchi, E. (2013) Genes 4(1):1-32
Back to the Top
Replication through Nucleosomes
Eukaryotic genomes are substantially more complicated than the smaller and unadorned prokaryotic genomes. Eukaryotic cells have multiple noncontiguous DNA components, chromosomes, each of which must be compacted to allow packaging within the confined space of a nucleus. As seen in chapter 4, chromosomes are packaged by wrapping ~147 nucleotides (at intervals averaging 200 nucleotides) around an octamer of histone proteins, forming the nucleosome. The histone octamer includes two copies each of histone H2A, H2B, H3, and H4. In chapter 8, it was highlighted that histone proteins are subject to a variety of post-translational modifications, including phosphorylation, acetylation, methylation, and ubiquitination that represent vital epigenetic marks. The tight association of histone proteins with DNA in nucleosomes suggests that eukaryotic cells possess proteins that are designed to remodel histones ahead of the replication fork, in order to allow smooth progression of the replisome. It is also essential to reassemble histones behind the fork to reestablish the nucleosome conformation. Furthermore, it is important to transmit the epigenetic information found on the parental nucleosomes to the daughter nucleosomes, in order to preserve the same chromatin state. In other words, the same histone modifications should be present on the daughter nucleosomes as were on the parental nucleosomes. This must all be done while doubling the amount of chromatin, which requires incorporation of newly synthesized histone proteins. This process is accomplished by histone chaperones and histone remodelers, which are discussed below (Figure 9.22).

Figure 9.22 Nucleosome displacement and deposition during DNA replication. Histones are removed from chromatin ahead of the replication fork. FACT may facilitate this process. Asf1 recruits histone H3-H4 dimers to the replication fork. CAF-1 and Rtt106 load newly synthesized (light purple) histones to establish chromatin behind the fork. Previously loaded histones (dark purple) are also deposited on both daughter DNA strands. The histone chaperones involved in these processes are associated with replisome proteins: CAF-1/Rtt106 with PCNA and FACT/Asf1 with MCMs.
Figure from: Lemanm A.R. and Noguchi, E. (2013) Genes 4(1):1-32
Several histone chaperones are known to be involved in replication-coupled nucleosome assembly, including the FACT complex. The FACT complex components were originally identified as proteins that greatly stimulate transcription by RNA polymerase II. In budding yeast, FACT was found to interact with DNA Pol α-primase complex, and the FACT subunits were found to interact genetically with replication factors. More recently, studies showed that FACT facilitates DNA replication in vivo and is associated with the replisome in budding yeast and human cells. The FACT complex is a heterodimer that does not hydrolyze ATP, but facilitates the “loosening” of histones in nucleosomes
Replication Fork Barriers and the Termination of Replication
In prokaryotes, such as the E. coli, bidirectional replication initiates at a single replication origin on the circular chromosome and terminates at a site approximately opposed from the origin. This replication terminator region contains DNA sequences known as Ter sites, polar replication terminators that are bound by the Tus protein. The Ter-Tus complex counteracts helicase activity, resulting in replication termination. In this way, prokaryotic replication forks pause and terminate in a predictable manner during each round of DNA replication.
In eukaryotes, the situation differs. Replication termination typically occurs by the collision of two replication forks anywhere between two active replication origins. The location of the collision can vary based on the replication rate of each of the forks and the timing of origin firing. Often, if a replication fork is stalled or collapsed at a specific site, replication of the site can be rescued when a replisome traveling in the opposite direction completes copying the region. However, there are numerous programmed replication fork barriers (RFBs) and replication “challenges” throughout the genome. To efficiently terminate or pause replication forks, some fork barriers are bound by RFB proteins in a manner analogous to E. coli Tus. In these circumstances, the replisome and the RFB proteins must specifically interact to stop replication fork progression.
Back to the Top
9.5 Replication of Mitochondrial DNA
Mammalian mitochondria contain multiple copies of a circular, double-stranded DNA genome approximately 16.6 kb in length (Figure 9.23). The two strands of mtDNA differ in their base composition, with one being rich in guanines, making it possible to separate a heavy (H) and a light (L) strand by density centrifugation.The mtDNA contains one longer noncoding region (NCR) also referred to as the control region. In the NCR, there are promoters for polycistronic transcription, one for each mtDNA strand; the light strand promoter (LSP) and the heavy strand promoter (HSP). The NCR also harbors the origin for H-strand DNA replication (OH). A second origin for L-strand DNA replication (OL) is located outside the NCR, within a tRNA cluster.

Figure 9.23 Map of human mtDNA. The genome encodes for 13 mRNA (green), 22 tRNA (violet), and 2 rRNA (pale blue) molecules. There is also a major noncoding region (NCR), which is shown enlarged at the top in blue. The major NCR contains the heavy strand promoter (HSP), the light strand promoter (LSP), three conserved sequence boxes (CSB1-3, orange), the H-strand origin of replication (OH), and the termination-associated sequence (TAS, yellow). The triple-stranded displacement-loop (D-loop) structure is formed by premature termination of nascent H-strand DNA synthesis at TAS. The short H-strand replication product formed in this manner is termed 7S DNA. A minor NCR, located approximately 11,000 bp downstream of OH, contains the L-strand origin of replication (OL).
Figure from: Falkenberg, M. (2018) Essays Biochem 62(3):287-296
A dedicated DNA replication machinery is required for its maintenance. Mammalian mtDNA is replicated by proteins distinct from those used for nuclear DNA replication and many are related to replication factors identified in bacteriophages. DNA polymerase γ (POLγ) is the replicative polymerase in mitochondria. In human cells, POLγ is a heterotrimer with one catalytic subunit (POLγA) and two accessory subunits (POLγB). POLγA belongs to the A family of DNA polymerases and contains a 3′–5′ exonuclease domain that acts to proofread the newly synthesized DNA strand. POLγ is a highly accurate DNA polymerase with a frequency of misincorporation lower than 1 × 10−6. The accessory POLγB subunit enhances interactions with the DNA template and increases both the catalytic activity and the processivity of POLγA. The DNA helicase TWINKLE travels in front of POLγ, unwinding the double-stranded DNA template. TWINKLE forms a hexamer and requires a fork structure (a single-stranded 5′-DNA loading site and a short 3′-tail) to load and initiate unwinding. Mitochondrial single-stranded DNA-binding protein (mtSSB) binds to the formed ssDNA, protects it against nucleases, and prevents secondary structure formation
The most accepted model of DNA replication in the mitochondria is the strand displacement model (Figure 9.24). Within this model, DNA synthesis is continuous on both the H- and L-strand. There is a dedicated origin for each strand, OH and OL. First, replication is initiated at OH and DNA synthesis then proceeds to produce a new H-strand. During the initial phase, there is no simultaneous L-strand synthesis and mtSSB covers the displaced, parental H-strand. By binding to single-stranded DNA, mtSSB prevents the mitochondrial RNA polymerase (POLRMT) from initiating random RNA synthesis on the displaced strand. When the replication fork has progressed about two-thirds of the genome, it passes the second origin of replication, OL. When exposed in its single-stranded conformation, the parental H-strand at OL folds into a stem–loop structure. The stem efficiently blocks mtSSB from binding and a short stretch of single-stranded DNA in the loop region therefore remains accessible, allowing POLRMT to initiate RNA synthesis. POLRMT is not processive on single-stranded DNA templates. After adding approximately 25 nucleotides, it is replaced by POLγ and L-strand DNA synthesis is initiated. From this point, H- and L-strand synthesis proceeds continuously until the two strands have reached full circle. Replication of the two strands is linked, since H-strand synthesis is required for initiation of L-strand synthesis. DNA Ligase III is used to complete the ligation of the newly formed DNA strands.
During DNA replication, the parental molecule remains intact, which poses a steric problem for the moving replication machinery. Topoisomerases belonging to the type 1 family can relieve torsional strain formed in this way, by allowing one of the strands to pass through the other. In mammalian mitochondria, TOP1MT a type IB enzyme can act as a DNA “swivel”, working together with the mitochondrial replisome. Furthermore, replication of circular DNA often causes the formation of catenanes, or interlocked circles that need to be separated from one another. The type 1A topoisomerase, topoisomerase 3α (Top3α), is required to resolve the hemicatenane structure that can form during mtDNA replication.

Figure 9.24 Replication of the human mitochondrial genome. Mitochondrial DNA replication is initiated at OH and proceeds unidirectionally to produce the full-length nascent H-strand. mtSSB binds and protects the exposed, parental H-strand. When the replisome passes OL, a stem–loop structure is formed that blocks mtSSB binding, presenting a single-stranded loop-region from which POLRMT can initiate primer synthesis. The transition to L-strand DNA synthesis takes place after about 25 nt, when POLγ replaces POLRMT at the 3′-end of the primer. Synthesis of the two strands proceeds in a continuous manner until two full, double-stranded DNA molecules have been formed.
Figure from: Falkenberg, M. (2018) Essays Biochem 62(3):287-296
Curiously, not all replication events initiated at OH continue to full circle. Instead, 95% are terminated after about the first 650 nucleotides at a sequence known as the termination associated sequences (TAS) (Figure 9.23). This creates a short DNA fragment known as the 7S DNA, that remains bound to the parental L-strand, while the parental H-strand is displaced (Figure 9.23). As a result, a triple-stranded displacement loop structure, a D-loop, is formed. The functional importance of the D-loop structure is unclear and how replication is terminated at TAS is also not known.
Back to the Top
9.6 Telomeres and Replicative Senescence
The End Replication Problem
In humans, telomeres consist of hundreds to thousands of repetitive sequences of TTAGGG at chromosomal ends for maintaining genomic integrity. Because the DNA replication is asymmetric along double strands, RNA pimer sequence at the 3′-hydroxyl end cannot be replaced by DNA polymerase I, as there is no 3′-OH primer group present for the polymerase to extend the DNA chain. This causes the loss of 30–200 nucleotides with each DNA replication and cell division and is known as the end replication problem. Telomeres provide a repetitive noncoding sequence of DNA at the 3′ end to prevent the loss of critical genetically encoded information during replication. Moreover, telomeres are coated with a complex of six capping proteins, also known as shelterin proteins, which are packed into a compact T-loop structure that hides the ends of the chromosomes. This prevents the DNA repair machinery from mistaking chromosomal ends for double-stranded DNA breaks (Figure 9.25). Therefore, telomeres have been proposed as a mitotic clock that measures how many times a cell has divided and in essence, gives a cell a defined lifetime.
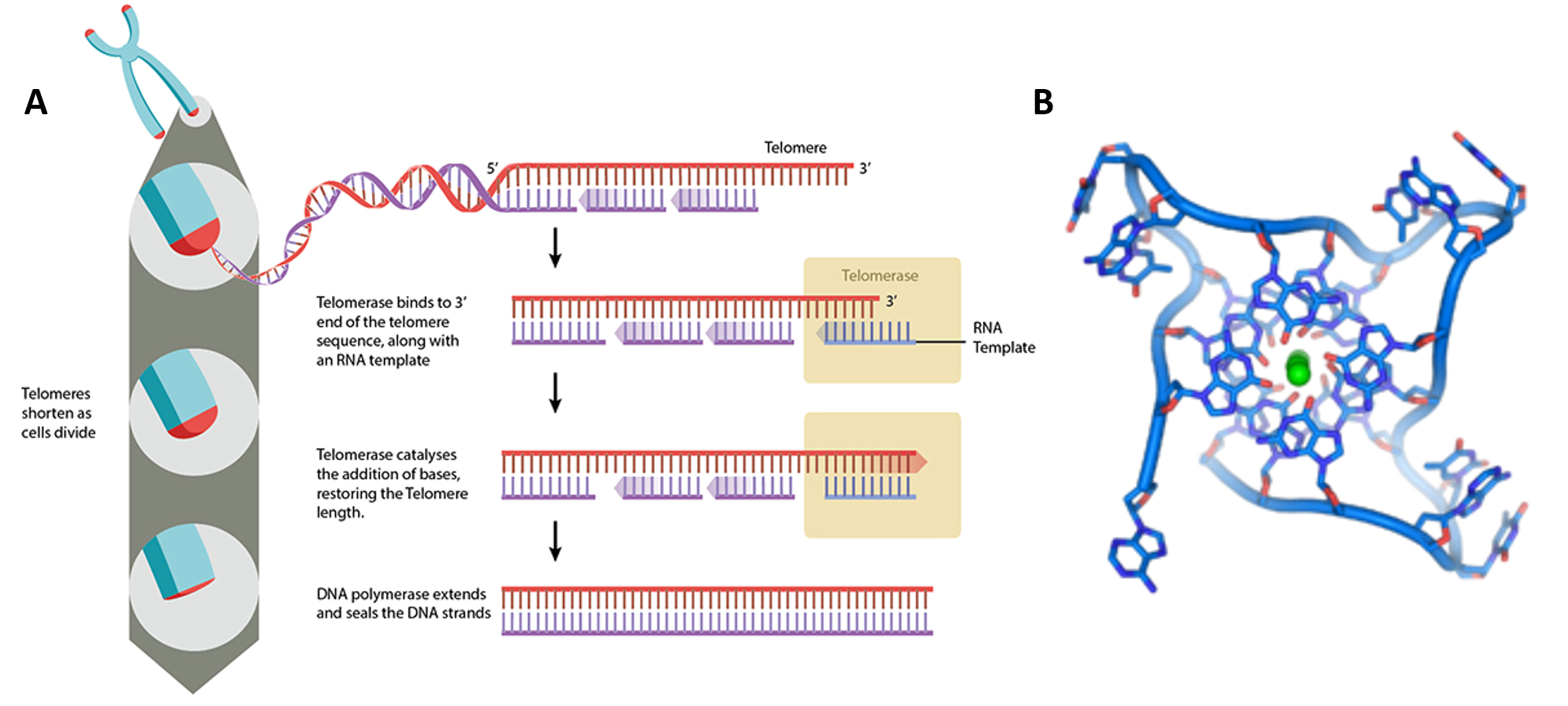
Figure 9.25 Telomere Structure. (A) Telomeres are located at the end of chromosomes, where they help protect against the loss of DNA during replication. (B) DNA quadruplex formed by telomere repeats. The looped conformation of the DNA backbone is very different from the typical DNA helix, this is known as T-loop formation. The green spheres in the center represent potassium ions.
Image (A) by: MBInfo and Image (B) by: Thomas Splettstoesser
{kind=link}
The human telomerase enzyme is responsible for maintaining and elongating telomeres and consists of an RNA component (TERC) and a reverse transcriptase (TERT), that serves as the catalytic component (Figure 9.26). The TERT uses the TERC as a template to synthesize new telomeric DNA repeats at a single-stranded overhang to maintain telomere length (Figure 9.26). Some cells such as germ cells, stem cells, hematopoietic progenitor cells, activated lymphocytes, and most cancer cells constitutively express telomerase and maintain telomerase activity to overcome telomere shortening and cellular senescence. However, most other somatic cells generally have a low or undetectable level of telomerase activity and concomitantly limited longevity. Interestingly, overall telomerase activity decreases with age, but increases markedly in response to injury, suggesting a role for telomerase in cellular regeneration during wound healing. The telomere length and integrity are regulated through the interplay between the telomerase and shelterin proteins.
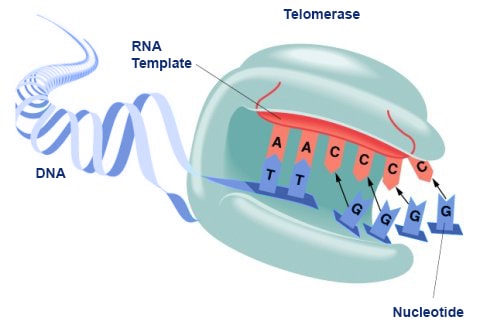
Figure 9.26 Conceptual Model of Telomerase Activity. The active site of the telomerase enzyme contains the RNA template, TERC (shown in red) and aligns with the last few telomeric bases at the end of the chromosome (shown in blue). This creates a single stranded overhang that can be used as a template by the TERT reverse transcriptase to extend the telomere sequence.
Figure by: Abbexa Ltd.
In vivo, shortened telomeres and damaged telomeres generally caused by reactive oxygen species (ROS) are usually assumed to be the main markers of cellular aging and are thought to be the main cause of replicative senescence. In vitro, telomeres loose approximately 50–200 bp at each division due to the end-replication problem. Approximately 100 mitoses are thought to be sufficient to reach the Hayflick limit, or the maximum number of mitotic events allowed prior to entering replicative senescence. Cells in continual renewal, such as blood cells, compensate for telomere erosion by expressing telomerase, the only enzyme able to polymerize telomeric sequences de novo at the extremity of telomeres. Knocking out telomerase components, such as the catalytic subunit (TERT) or the RNA template (TERC), induces several features of aging in mice. In humans, germline mutations in telomerase subunits are responsible for progeroïd syndromes, such as Dyskeratosis congenita, a rare genetic form of bone marrow failure. Furthermore, healthy lifespan in humans is positively correlated with longer telomere length and patients suffering from age-related diseases and premature aging have shorter telomeres compared with healthy individuals. An accumulation of unrepaired damage within telomeric regions has also been shown to accumulate in aging mice and non-human primates, suggesting that damage of telomeres with age may also be contributing to age-driven disease states and reduced healthspan.
Thus, one could argue that the activation and expression of telomerase may be a way of reducing age-related diseases and increasing overall longevity. However, the constitutive expression of telomerase, unfortunately, is a characteristic of almost all cancer cells. It is therefore, no surprise that transgenic animals over-expressiong the catalytic subunit of telomerase (mTERT), develop cancers earlier in life. However, overexpression of telomerase in mice that are highly resistant to cancers has shown large increases in median lifespan and significantly reduced age-associated disorders. Since humans are not highly resistant to cancer, this is not a feasible option for humans. However, additional studies in mice, where constitutive expression of telomerase is only introduced into a small percentage of host cells using adenovirus gene therapy techniques has yielded more promising results. Adenoviruses are a group of viruses that form an icosahedral protein capsid that houses a linear double stranded DNA genome. Infections in humans typically cause symptoms of the common cold and are usually mild in nature. These are a good target for gene therapy, as the DNA that they carry can be mutated, so that they are deficient in their ability to replicate once they have infected the host. They can also be transformed to carry a gene-of-interest into the host, where that gene can then integrate into the host genome. Experiments in mice that were infected with an adenovirus carrying the mTERT gene showed that mTERT was delivered to a wide range of tissues within the body, and increased telomere length within those tissues. Furthermore, the mTERT expressing mice were healthier than their litter mates and displayed a reduction in disabling conditions associated with physiological aging such as osteoporosis and insulin resistance (Figure 9.27). Cognitive skills and metabolic functions were also improved. Noticeably, mice treated with gene therapy did not have increased incidence in cancer rates, suggesting that in at least the short-lived mouse species, that a gene therapy approach to increased telomerase activity is safe. Within these animals, median lifespan was increased by 24% when animals were treated at 1 year of age, and by 13% if treated at 2 years of age.
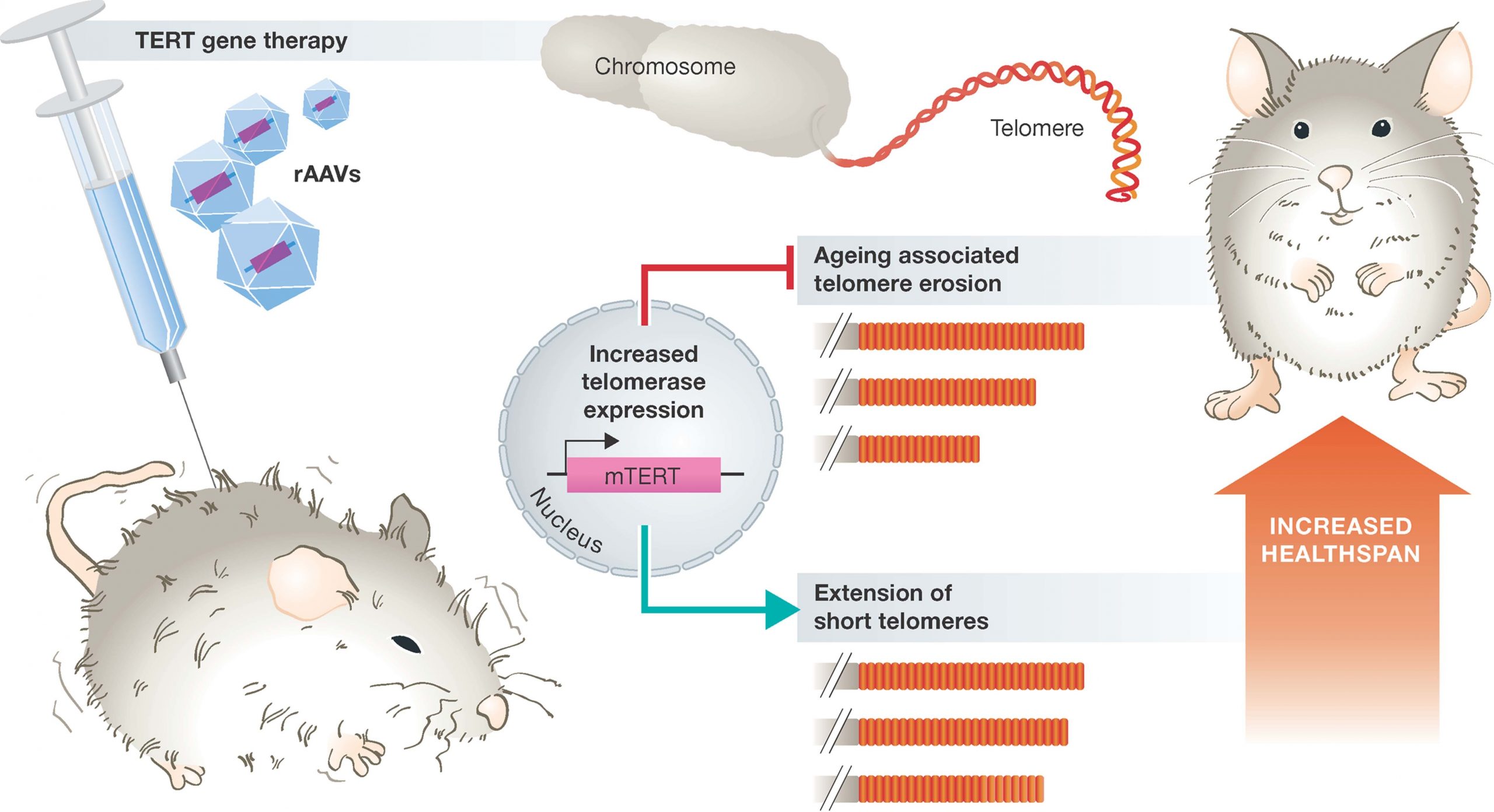
Figure 9.27 Promoting Healthspan in Mice using a Telomerase Gene Therapy. Delivery of the catalytic subunit of telomerase (TERT) using a modified adenovirus vector (rAAV) suppresses aging associated telomere erosion and extends short telomoeres in a variety of tissues. Consequently, animals display improved healthspan and extended lifespan.
Figure from: Boccardi, V. and Herbig, U. (2012) EMBO Mol Med 4:685-687.
Replication and Repair of Telomere Sequences
In addition to the end replication problem, telomeric DNA (telDNA) replication and repair is a real challenge due to the different structural features of telomeres. Firstly, the nucleotidic sequence itself consists of an hexanucleotidic motif (TTAGGG) repeated over kilobases, with the 5′-3′ strand named the “G-strand” due to its high content in guanine. During the progression of the replication fork, the lagging strand, corresponding to the G-strand, forms G-quadruplex (G4) structures, which have to be resolved to allow fork progression and to complete replication (Figure 9.28a). Secondly, R-loops corresponding to highly stable RNA:DNA hybrids, involving the long non-coding telomeric transcript TERRA (telomeric repeat-containing RNA) also have to be dissociated. Thirdly, the extremity of telomeres adopts a specific loop structure, the T-loop, which has to be unraveled. This is the loop that hides the double strand end from the DNA damage sensors, and is locked by the hybridization of the 3′ single strand overhang extremity with the above 3′-5′ strand, thereby displacing the corresponding 5′-3′ strand to form a D-loop (displacement loop) structure (Figure 9.28a). Lastly, replication also has to deal with barriers encountered elsewhere in the genome, such as torsions and a condensed heterochromatic environment.
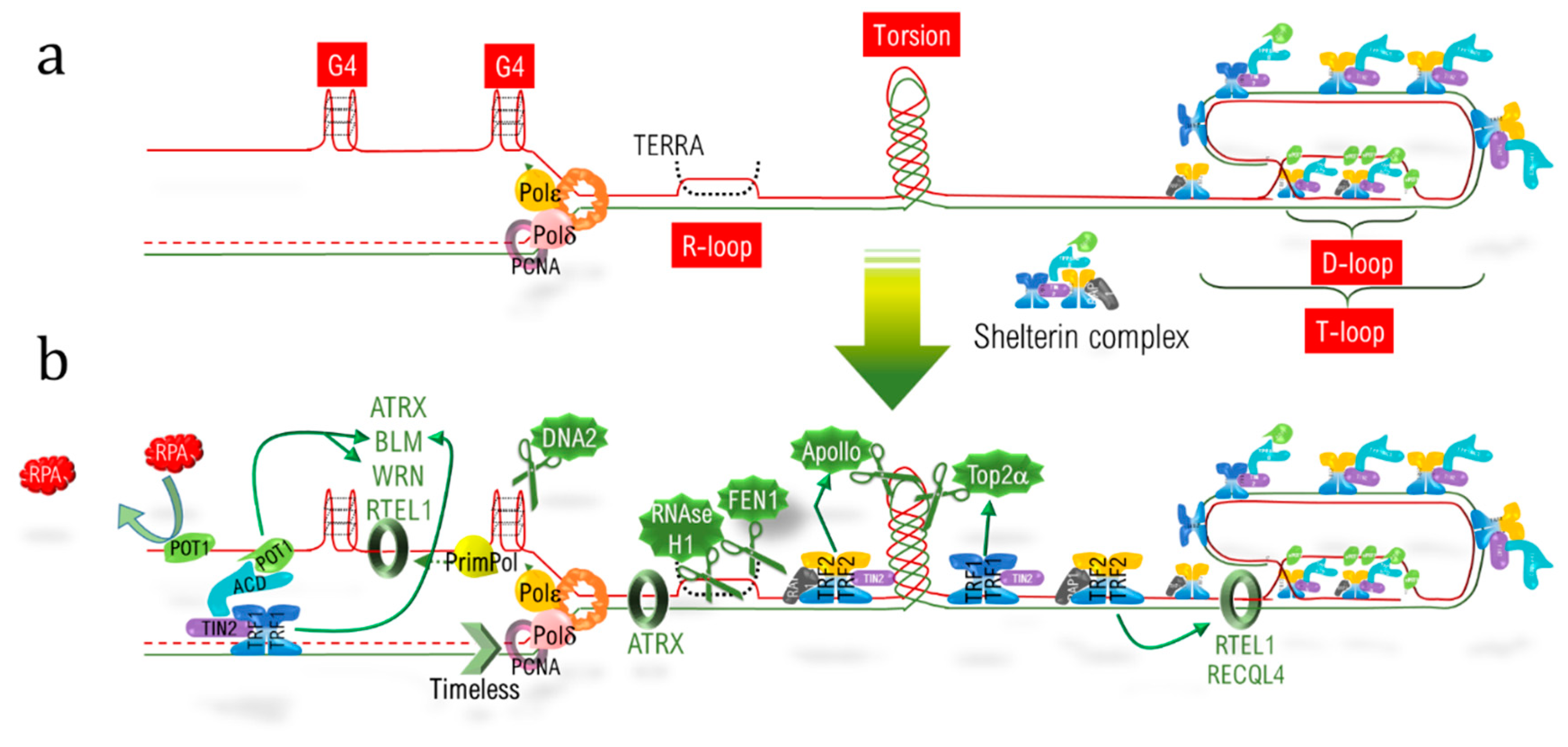
Figure 9.28 Obstacles and solutions to replicate telomeres. (a) Telomeric sequence, with the G-strand in solid red line and the C-strand in solid green line, is depicted. The terminal D-loop structuring the much larger T-loop is stabilized by the shelterin complex. The replisome (PCNA, pol ε, etc) polymerizes a new G-strand (depicted in dotted red line) and frees the parental G-strand, enabling the formation of G4 secondary structure. R-loops corresponding to TERRA hybridization (in dotted black lines) with the 3′-5′ strand, and torsions due to the fork progression are also shown. (b) Replication helpers, such as helicases, either helping in G4 unwinding or in D-loop unlocking are depicted. The DNAses (Top2a, DNA2) and RNAses (RNAse H1 and FEN1) help in resolving torsions and RNA:DNA heteroduplexes, while Timeless stimulates the replisome and POT1 competes with RPA1 for binding of the single-strand and helps in G4 dissolution. The shelterin components, POT1, TRF1 and TRF2 help in loading the helper-proteins (fine green arrows)
Figure from: Billiard, P. and Poncet, D.A. (2019) Int J. Mol. Sci. 20(19) 4959
Since telomeres face a host of obstacles to completing the replication process, as discussed in Figure 9.28, the cell possess a set of specialized machinery to fully achieve their replication, such as the RTEL1, TRF1, and TRF2 proteins, DNAses, RNAsses, and Timeless. The recruitment of these factors is orchestrated by the shelterin complex.
At the molecular level, the GGG telomeric repeats are particularly sensitive to ROS, which produce stretches of 8-oxoguanine that are especially difficult to repair. Coupled with inefficient telomere repair, these ROS-induced lesions produce single and double-strand breaks, and/or generate replicative stress, ultimately resulting in telomere shortening. The presence of unrepaired single or tandem 8-oxoguanine drastically inhibits the binding of TRF1 and TRF2, and impairs the recruitment of telomerase, especially when ROS damage is localized in the 3′ overhang. This type of damage contributes to telomere deprotection and shortening. Strikingly, ROS (and other metabolic stresses) also induce the relocation of TERT to mitochondria, as observed (i) in primary neurons after oxidative stress; (ii) in neurons exposed to the tau protein; (iii) in Purkinje neurons subjected to excitotoxicity; and (iv) in cancer cell lines treated with a G4 ligand. Mitochondrial TERT increases the inner membrane potential, as well as the mtDNA copy number, and decreases ROS production with a protective effect on mtDNA. Mitochondria are also critical sensors of cellular damage and contribute to the processes of autophagy and apoptosis (programmed cell death). The relocalization of TERT following chromosomal damage in the nucleus, may indicate one mechanism the mitochondria utilizes to monitor cellular stress and damage.
Back to the Top
Mastering the Content
Which of the following is the enzyme that replaces the RNA nucleotides in a primer with DNA nucleotides?
- DNA polymerase III
- DNA polymerase I
- primase
- helicase
[reveal-answer q=”628075″]Show Answer[/reveal-answer]
[hidden-answer a=”628075″]Answer b. DNA polymerase I is the enzyme that replaces the RNA nucleotides in a primer with DNA nucleotides.[/hidden-answer]
Which of the following is not involved in the initiation of replication?
- ligase
- DNA gyrase
- single-stranded binding protein
- primase
[reveal-answer q=”820951″]Show Answer[/reveal-answer]
[hidden-answer a=”820951″]Answer a. Ligase is not involved in the initiation of replication.[/hidden-answer]
Which of the following enzymes involved in DNA replication is unique to eukaryotes?
- helicase
- DNA polymerase
- ligase
- telomerase
[reveal-answer q=”650146″]Show Answer[/reveal-answer]
[hidden-answer a=”650146″]Answer d. Telomerase is unique to eukaryotes.[/hidden-answer]
Which of the following would be synthesized using 5′-CAGTTCGGA-3′ as a template?
- 3′-AGGCTTGAC-4′
- 3′-TCCGAACTG-5′
- 3′-GTCAAGCCT-5′
- 3′-CAGTTCGGA-5′
[reveal-answer q=”429167″]Show Answer[/reveal-answer]
[hidden-answer a=”429167″]Answer c. 3′-GTCAAGCCT-5′[/hidden-answer]
The enzyme responsible for relaxing supercoiled DNA to allow for the initiation of replication is called ________.
[reveal-answer q=”855893″]Show Answer[/reveal-answer]
[hidden-answer a=”855893″]The enzyme responsible for relaxing supercoiled DNA to allow for the initiation of replication is calledDNA gyrase or topoisomerase II.[/hidden-answer]
Unidirectional replication of a circular DNA molecule like a plasmid that involves nicking one DNA strand and displacing it while synthesizing a new strand is called ________.
[reveal-answer q=”378861″]Show Answer[/reveal-answer]
[hidden-answer a=”378861″]Unidirectional replication of a circular DNA molecule like a plasmid that involves nicking one DNA strand and displacing it while synthesizing a new strand is calledrolling circle replication.[/hidden-answer]
More primers are used in lagging strand synthesis than in leading strand synthesis.
[reveal-answer q=”25479″]Show Answer[/reveal-answer]
[hidden-answer a=”25479″]True[/hidden-answer]
- Why is primase required for DNA replication?
- What is the role of single-stranded binding protein in DNA replication?
- Below is a DNA sequence. Envision that this is a section of a DNA molecule that has separated in preparation for replication, so you are only seeing one DNA strand. Construct the complementary DNA sequence (indicating 5′ and 3′ ends).DNA sequence: 3′-T A C T G A C T G A C G A T C-5′
- Review Figure 1 and Figure 2. Why was it important that Meselson and Stahl continue their experiment to at least two rounds of replication after isotopic labeling of the starting DNA with15N, instead of stopping the experiment after only one round of replication?
- If deoxyribonucleotides that lack the 3′-OH groups are added during the replication process, what do you expect will occur?
9.7 References
- Parker, N., Schneegurt, M., Thi Tu, A-H., Lister, P., Forster, B.M. (2019) Microbiology. Openstax. Available at: https://opentextbc.ca/microbiologyopenstax/
- Principles of Biochemistry/Cell Metabolism I: DNA replication. (2017, August 6). Wikibooks, The Free Textbook Project. Retrieved 19:07, October 31, 2019 from https://en.wikibooks.org/w/index.php?title=Principles_of_Biochemistry/Cell_Metabolism_I:_DNA_replication&oldid=3259729.
- Kaiser, G.E. (2015) Prokaryotic Cell Anatomy. Community College of Baltimore County. Available at: http://faculty.ccbcmd.edu/~gkaiser/SoftChalk%20BIOL%20230/Prokaryotic%20Cell%20Anatomy/nucleoid/nucleoid/nucleoid3.html
- The RCSB PDB “Molecule of the Month”: Inspiring a Molecular View of Biology D.S. Goodsell, S. Dutta, C. Zardecki, M. Voigt, H.M. Berman, S.K. Burley (2015) PLoS Biol 13(5): e1002140. doi: 10.1371/journal.pbio.1002140
- Wikipedia contributors. (2020, May 7). Helicase. In Wikipedia, The Free Encyclopedia. Retrieved 13:38, June 9, 2020, from https://en.wikipedia.org/w/index.php?title=Helicase&oldid=955303097
- Windgassen, T.A., Wessel, S.R., Bhattacharyya, B., and Keck, J.L. (2017) Mechanisms of bacterial DNA replication restart. Nuc Acids Res 46(2):504-519. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5778457/
- Xu, Z-Q., Dixon, N.E. (2018) Bacterial Replisomes. Curr Opin Struct Biol 53:159-168. Available at: https://www.sciencedirect.com/science/article/pii/S0959440X18300952
- Liu, B., Eliason, W.K., and Steitz, T.A. (2013) Structure of a helicase-helicase loader complex reveals insights into the mechanism of bacterial primosome assembly. Nature Comm 4:2495. Available at: https://www.researchgate.net/publication/256764134_Structure_of_a_helicase-helicase_loader_complex_reveals_insights_into_the_mechanism_of_bacterial_primosome_assembly
- Xu, Z-Q., and Dixon, N.E. (2018) Bacterial Replisomes. Curr Op Struc Biol 53:159-168. Available at: https://www.sciencedirect.com/science/article/pii/S0959440X18300952
- Fernandez-Leiro, R., Conrad, J., Scheres, S.HW., and Lamers, M.H. (2015) cryo-EM structures of the E. coli replicative DNA polymerase reveal its dynamic interactions with the DNA sliding clamp, exonuclease and τ. eLife 4:e11134. Available at: https://elifesciences.org/articles/11134
- Ekundayo, B. and Bleichert, F. (2019) Origins of DNA replication. PLOS 15(12): e1008556. Available at: https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1008320
- Leman, A.R., and Noguchi, E. (2013) The replication fork: understanding the eukaryotic replication machinery and the challenges to genome duplication. Genes 4(1): 1-32. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3627427/
- Doublié, S. and Zahn, K.E. (2014) Structural insights into eukaryotic DNA replication. Front. Microbiol. 5:444. Available at: https://www.frontiersin.org/articles/10.3389/fmicb.2014.00444/full
- Billard, P., and Poncet D.A. (2019) Replication stress at telomeric and mitochondrial DNA: Common origins and consequences on ageing. Int J. Mol Sci 20(19):4959. Available at: https://www.mdpi.com/1422-0067/20/19/4959/htm
- Yeh, J-K., and Wang, C-Y. (2016) Telomeres and telomerase in cardiovascular diseases. Genes 7(9)58. Available at: https://www.mdpi.com/2073-4425/7/9/58/htm
- Boccardi, V. and Herbig, U. (2012) Telomerse gene therapy: a novel approach to combat aging. EMBO Mol Med 4:685-687. Available at: https://www.embopress.org/doi/epdf/10.1002/emmm.201200246
- Wikipedia contributors. (2020, April 26). Cyclin-dependent kinase. In Wikipedia, The Free Encyclopedia. Retrieved 18:52, June 30, 2020, from https://en.wikipedia.org/w/index.php?title=Cyclin-dependent_kinase&oldid=953307433
- Falkenberg, M. (2018) Mitochondrial DNA replication in mammalian cells: overview of the pathway. Essays Biochem 62(3):287-296. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6056714/
- Folwer, S., et. al. (2013) Concepts of Biology. Openstax. Available at: https://openstax.org/details/books/concepts-biology?Book%20details
- Aleem, E. and Arceci, R.J. (2015) Targeting cell cycle regulators in hematologic malignancies. Frontiers in Cell and Developmental Biology 3(16). Available at: https://www.researchgate.net/publication/275354547_Targeting_cell_cycle_regulators_in_hematologic_malignancies