Home » Student Resources » Online Chemistry Textbooks » CH450 and CH451: Biochemistry - Defining Life at the Molecular Level » Chapter 8 – Protein Regulation and Degradation
MenuCH450 and CH451: Biochemistry - Defining Life at the Molecular Level
Chapter 8 – Protein Regulation and Degradation
8.1 Isozymes
8.2 Post-Translational Modifcations
8.3 Allosteric Regulation
8.4 Zymogen Activation
8.5 Intracellular Protein Degradation
8.6 References
Protein activity within cells is controlled by many different mechanisms. The primary sequence of a protein is a main determinant of protein folding and final conformation as well as biochemical activity, stability, and half-life. However, at any given moment in the life of an individual, its proteome is up to two or three orders of magnitude more complex than the encoding genomes would predict. This chapter gives an overview of the major mechanisms utilized by biological systems to regulate protein functions after the protein has been synthesized. Note that these mechanisms seldom work in isolation. There are usually multiple levels of protein control that are functioning at any given time and in response to many different environmental cues and signals.
8.1 Isozymes
Isozymes (also known as isoenzymes or more generally as multiple forms of enzymes) are enzymes that differ in amino acid sequence but catalyze the same chemical reaction. These enzymes usually display different kinetic parameters (e.g. different KM or Kcat values), or different regulatory properties. The existence of isozymes permits the fine-tuning of metabolism to meet the particular needs of a given tissue or developmental stage. In many cases, isozymes are coded for by homologous genes that have been duplicated within the genome and have then diverged over time. Isozymes should not be confused with allozymes, which are allelic variants of the same gene locus that are found within a population. Allozymes represent enzymes from different alleles of the same gene, whereas isozymes represent enzymes from different genes that process or catalyse the same reaction, the two words should not be used interchangeably. We will focus on isozymes within this section.
Isozymes are usually the result of gene duplication. Over evolutionary time, if the function of the new variant remains identical to the original, then it is likely that one or the other will be lost as mutations accumulate, resulting in a pseudogene. However, if the mutations do not immediately prevent the enzyme from functioning, but instead modify either its function, or its pattern of expression, then the two variants may both be favored by natural selection and become specialized to different functions. For example, they may be expressed at different stages of development or in different tissues. Some isozymes may also arise from convergent evolution and may not share high sequence homology or common ancestry.
The Cyclooxygenase Enzymes, Cox-1 and Cox-2 are Isozymes
The Cyclooxygenases COX-1 and COX-2, also called Prostaglandin Synthases, are examples of isoenzymes. They regulate a key step in prostaglandin and thromboxane synthesis and are the targets of nonsteroidal antiinflammatory drugs (NSAIDs), such as ibuprofen (Figure 8.1). The prostaglandins (PG) are a group of physiologically active lipid compounds called eicosanoids having diverse hormone-like effects in animals. Prostaglandins have been found in almost every tissue in humans and other animals. They are derived enzymatically from the fatty acid arachidonic acid. Every prostaglandin contains 20 carbon atoms, including a 5-carbon ring. They are a subclass of eicosanoids and of the prostanoid class of fatty acid derivatives.
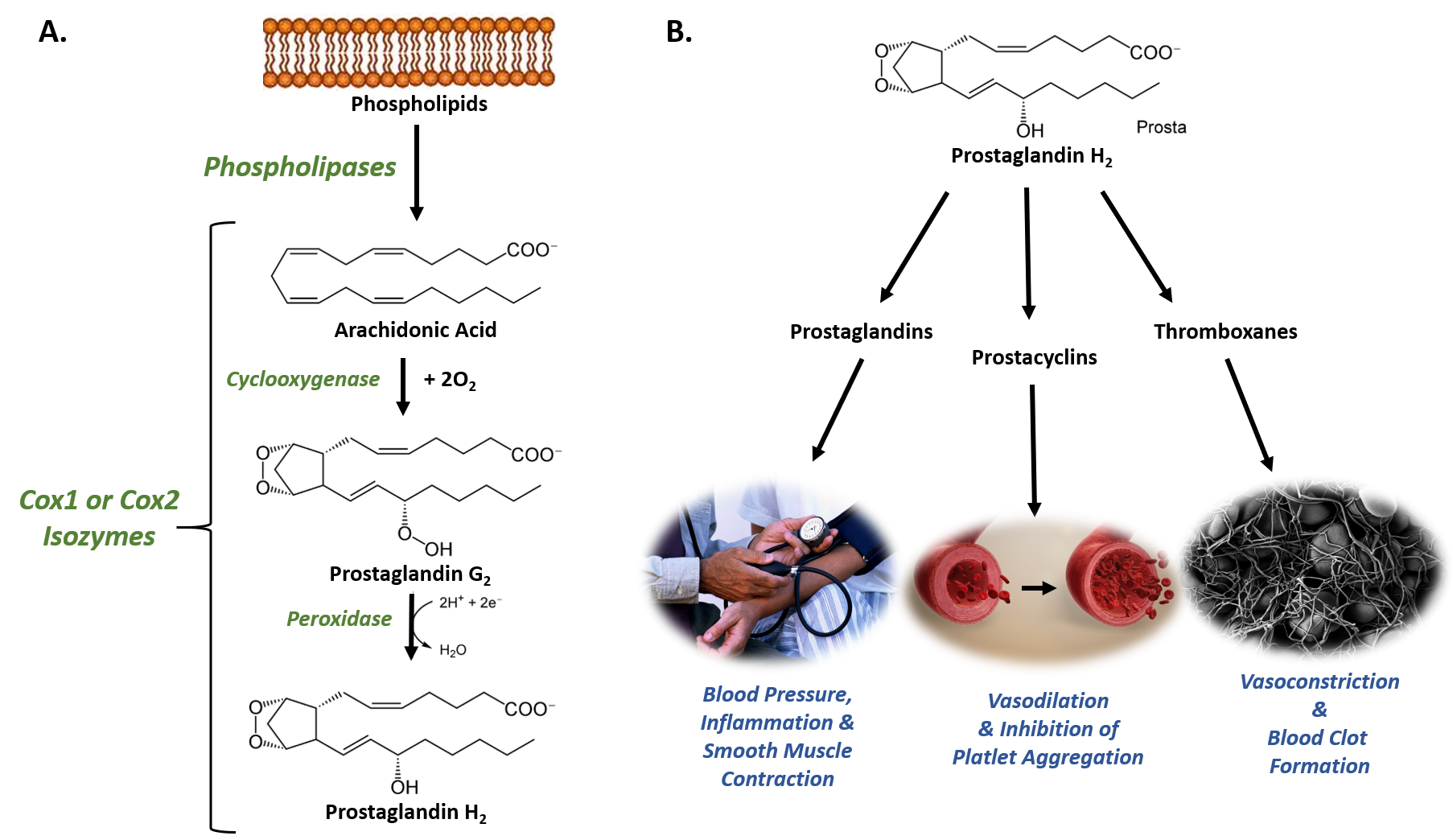
Figure 8.1 The Role of COX-1 and COX-2 in Prostaglandin Biosynthesis. (A) COX-1 and COX-2 are bifunctional enzymes that mediate the cyclooxygenase and peroxidase reactions that convert Arachidonic Acid to Prostaglandin H2. (B) Some of the physiological effects that prostaglandins, prostacyclins, and thromboxanes have on biological processes.
Images from: Администрация Волгоградской области , scientific animations, and Fuzis
The structural differences between prostaglandins account for their different biological activities. A given prostaglandin may have different and even opposite effects in different tissues. The ability of the same prostaglandin to stimulate a reaction in one tissue and inhibit the same reaction in another tissue is determined by the type of receptor to which the prostaglandin binds. They act as autocrine or paracrine factors with their target cells present in the immediate vicinity of the site of their secretion. Prostaglandins differ from endocrine hormones in that they are not produced at a specific site but in many places throughout the human body and tend to act locally once secreted. Prostaglandins are implicated in various physiological processes such as gastrointestinal cytoprotection, hemostasis and thrombosis, as well as renal hemodynamics.
Through their role in vasodilation, prostaglandins are also involved in inflammation and can trigger the onset of a fever or the sensation of pain. They are synthesized in the walls of blood vessels and serve the physiological function of preventing needless clot formation, as well as regulating the contraction of smooth muscle tissue. The prostacyclins, a special class of prostaglandins, are powerful, locally-acting vasodilators and inhibit the aggregation of blood platelets. Conversely, thromboxanes (produced by platelet cells) are vasoconstrictors and facilitate platelet aggregation. Their name comes from their role in clot formation or thrombosis (Figure 8.1).
The Cyclooxygenases COX-1 and COX-2 regulate the first two steps in prostaglandin and are bifunctional enzymes containing two active sites. The first active site performs the bis-oxygenation and cyclization of arachidonic acid, whereas the second active site mediates a peroxidase reaction to form PGH2 (Figure 8.1). The COX-1 enzyme is widely distributed in many tissues where it is constitutively expressed. Expression of the COX-2 isoform (shown in Figure 8.2), on the otherhand, is normally undetectable in most tissues (except for the central nervous system, kidneys, and seminal vesicles). COX-2 is induced by various inflammatory and mitogenic stimuli. More recently, a third isoform named COX-3 was identified as a COX-1 splicing variant. This new variant may play a role in processes such as fever and pain. Additionally, a high level of COX-2 expression is found in several forms of cancer. For example, COX-2 overexpression is related to poor prognosis in certain breast cancers and endometrial adenocarcinomas.
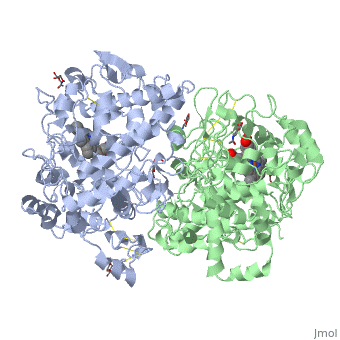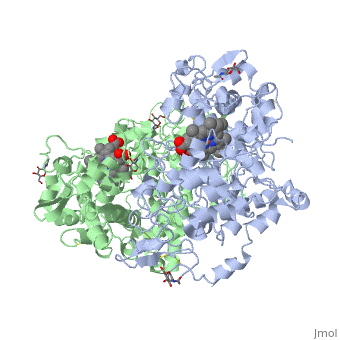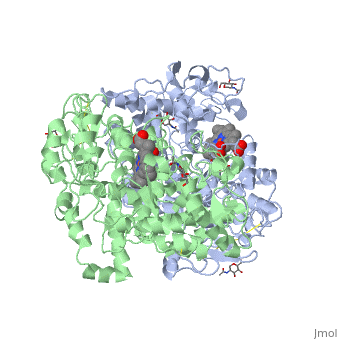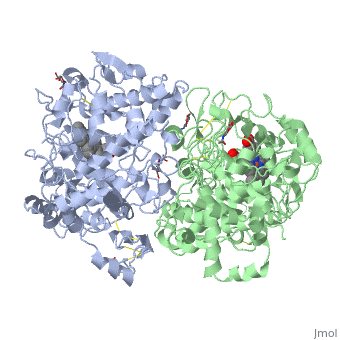
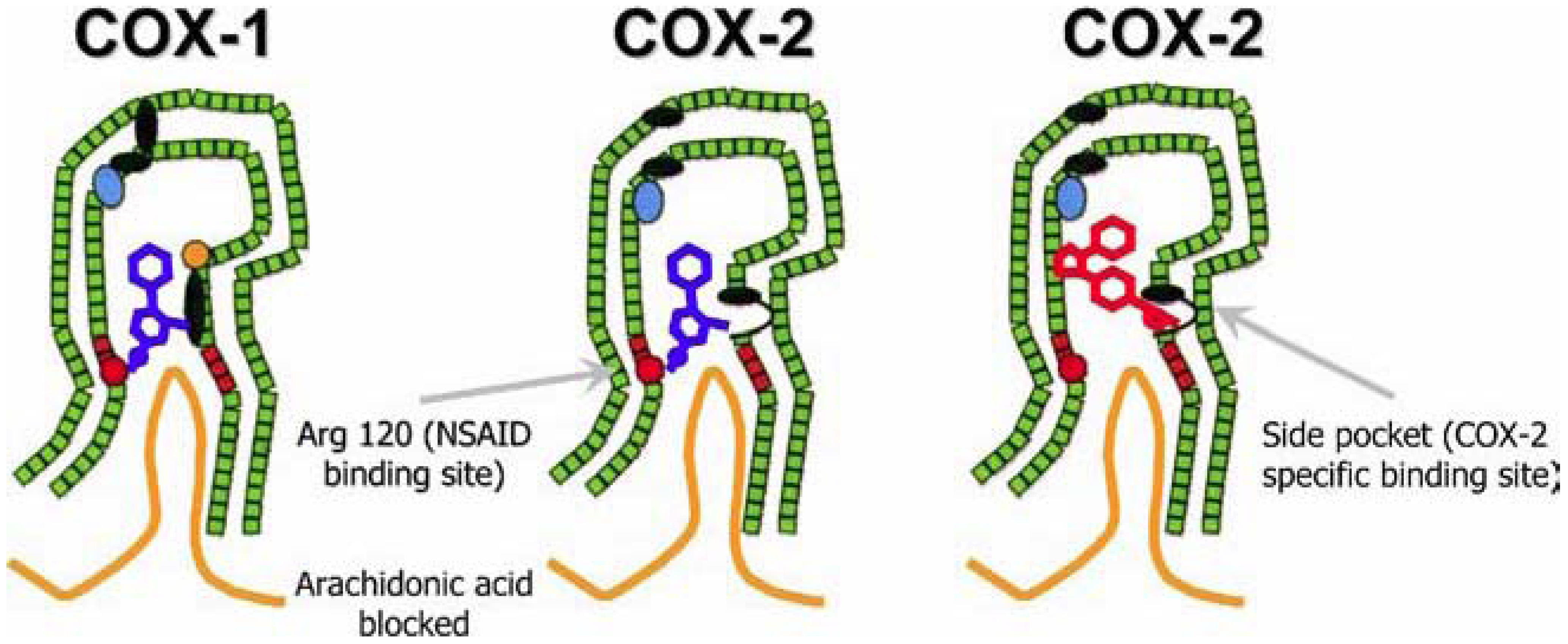
Figure 8.2 NSAID Inhibition of COX-1 and COX-2 Enzymes. Upper panel shows the crystal structure of the glycosylated, mouse COX-2 tetramer with heme group cofactor shown in red and gray. Lower left panel shows the schematic representation of the COX-1 (large green figure) active site being inhibitied by a nonselective NSAID (central blue figure). The entrance channel to COX-1 is blocked by the NSAID. Binding and transformation of arachidonic acid (bottom yellow figure) within COX-1 is prevented. Middle Lower Panel shows the inhibition of COX-2 by a nonselective NSAID (central blue figure). Nonselective binding uses an amino acid residue, Arg120, that is conserved in both enzymes. Lower right panel shows the inhibition of COX-2 by COX-2 selective NSAIDs (central red figure). The COX-2 side pocket allows specific binding of the COX-2 selective NSAID’s. The entrance channel to COX-2 is blocked. The bulkier COX-2-selective NSAID will not fit into the narrower COX-1 entrance channel, allowing COX-1 to remain active.
Upper Figure by Saiz, M., Gonzalez, R., & Garcia, E., (2019) Protopedia and Lower Figure by Meek, I.L., et al. (2010) Pharmaceuticals 3(7):2146-2162.
back to the top
COX-2, unlike COX-1, is induced in inflammatory cells when they are activated by various inflammatory and mitogenic stimuli. Under these conditions, COX-2 activity leads to the production of prostanoid mediators that trigger important inflammatory processes. Although inflammation is initially a necessary process to fight infection or build up an efficacious inmmune response, if it is maintained or remains uncontrolled, it can provoke chronic pathologies and tissue damage. This is why the inhibition of COX proteins have created considerable interest as potent anti-inflammatory and pain-management targets and has resulted in the development and use of NSAIDs (Figure 8.2).
Clinically available NSAIDs can be separated into 3 different classes based upon their mechanism of action:
-
ASPIRIN: – Acts to irreversibly inhibit COX 1 & COX-2 by covalent acetylation of serine residues in their respective active sites. Most notably, low doses of aspirin can suppress platelet COX-1 activity by 95% or more, an effect that is permanent for the lifetime of the platelet, since platelets lack DNA and cannot synthesize new enzyme. Due to aspirin’s antithrombotic property at low doses, this treatment has been found to have cardioprotective effects and is often prescribed for patients at high risk of myocardial infarction. All other NSAIDs interact with COX isoforms reversibly and produce variable COX inhibition (ranging from 50% to 95%) in a time-dependent fashion based upon their pharmacokinetic properties.
- NON-SELECTIVE COX INHIBITORS: Different non-selective NSAIDs have varying inhibitory effects against COX-1 & COX-2 (Figure 8.3). The two most commonly used over-the-counter drugs in this group (ibuprofen & naproxen) produce reversible platelet inhibition ranging from 50 to 95% in a reversible, time-dependent manner. These NSAIDs may be insufficient to provide cardio-protection throughout a commonly used dosing interval and are not commonly used for this purpose. Ketorolac (Toradol ®), an NSAID most commonly used in a hospital setting to treat moderately severe pain, is classified as a non-selective NSAID, although it is arguably, a very selective for COX-1 inhibitor (Figure 8.3). Inhibition of COX-1 can result in unwanted side effects, such as gastrointestinal discomfort and in severe cases, ulceration.
-
COXIBS: Selective COX-2 inhibitors were designed and marketed to treat pain and inflammation, while avoiding the GI side effects known to result from suppression protective prostaglandins synthesized by COX-1 in the GI mucosa. However soon after they were introduced into the marked, their use led to the first reported incidence of increased cardiovascular events (myocardial infarction and stroke) in 2004. Rofecoxib (Vioxx ®), one of the most selective COX-2 inhibitors was removed from the market because of mounting evidence for significant cardiovascular toxicity (Drazen, 2005). Celecoxib (Celebrex ®) is currently the only FDA approved coxib available in the US, and it has been given a black box warning indicating the potential risk of cardiovascular toxicity. It has a 10-20 fold selectivity for COX-2 over COX-1. Etoricoxib (Arcoxia ®) is a second coxib with ~106 fold selectivity for COX-2 over COX-1 that is available outside of the United States.
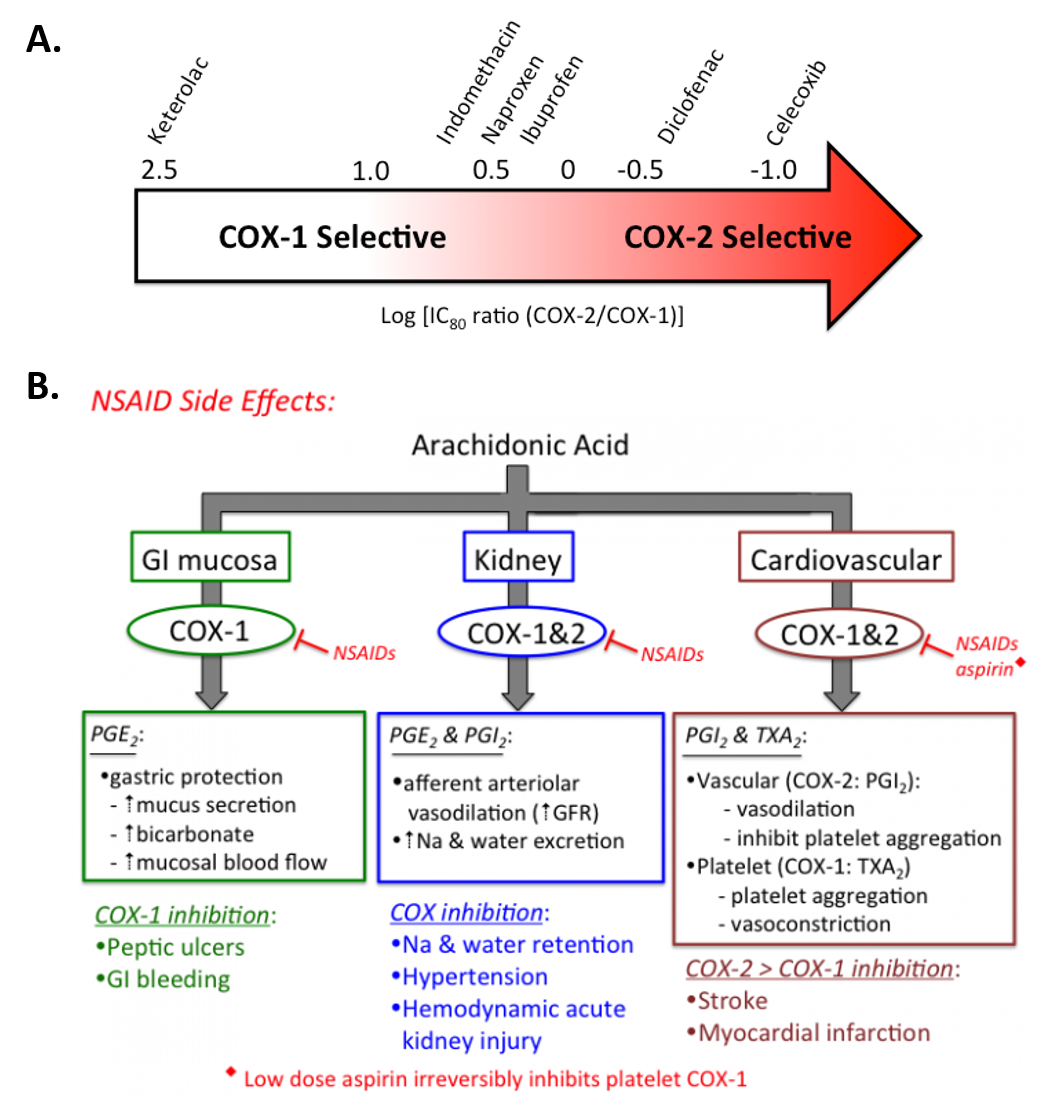
Figure 8.3 Selectivity and Treatment Efficacy of Nonsteroidal Anti-inflammatory Drugs (NSAIDs). (A) Relative COX-1 & COX-2 selectivity for commonly used non-aspirin NSAIDs. Celecoxib (Celebrex ®) is the only COX-2 selective NSAID on the market in the US. Adapted from Danelich et al (2015). (B) Major physiological roles for COX-1 & COX-2, and mechanisms underlying drug-induced side effects. PGI2: prostacyclin, TXA2: thromboxane.
Figures from: Clarkson, C.W. (2018) TUSOM Pharmwiki
Coxibs and the Thromboxane/Prostacyclin Imbalance Hypothesis
Previous research indicates that in the cardiovascular system, a greater inhibition of COX-2 vs COX-1 (as produced by COX-2 selective “coxibs”) can tip the normal balance between the effects produced by prostacyclin & thromboxane, resulting in an increased likelihood for platelet aggregation and vasoconstriction. These effects can help to explain the higher incidence of myocardial infarction and stroke observed when these drugs have been used clinically. The mechanisms involved are illustrated in Figure 8.4.

Figure 8.4. COX-2 Inhibitors & Cardiovascular Risk. The left graphic illustrates the normal balanced effect between prostacyclin (PGI2) and Thromboxane (TXA2). PGI2 is produced primarily by COX-2 activity in the endothelial cell wall of blood vessels. PGI2 produces vasodilation, and inhibits platelet activation. In contrast, TXA2 is produced primarily by COX-1 activity inside platelets, and produces vasoconstriction and enhanced platelet aggregation. When their is a balanced effect of both PGI2 & TXA2, normal vascular homeostasis is maintained. However, when the balance is tipped in favor of TXA2 formation after selective inhibition of COX-2 (right graphic), vasoconstriction and platelet clumping are more likely to occur, potentially causing an increased risk for cardiovascular events such as myocardial infarction and stroke.
Figure from: Clarkson, C.W. (2018) TUSOM Pharmwiki
Overall, the COX-1/COX-2 isozyme example sheds light on the complexity of biological systems and the ability for slight adjustments in gene expression to create varied and tissue-specific responses. We will revisit the complexity of isoform variability in Chapters X – XX, in our discussions about carbohydrate metabolism.
back to the top
8.2 Post-Translational Modifcations
The human genome contains approximately 20,000 to 25,000 genes. When analyzing the transcriptome, it becomes apparent that the genome becomes amplified by the wide array of splice vairants that can occur during the processing of transcripts. It is estimated that the transcriptome contains roughly 100,000 transcripts. This is amplified again within the proteome that contains over 1,000,000 unique proteins. One of the main routes of proteome expansion is through posttranslational modifications (PTM) of proteins. PTMs are present in both eukaryotes and prokaryotes, but it is estimated that PTMs are more common in eukaryotic cells, in which about 5% of the genome is dedicated to enzymes that carry out posttranslational modifications of proteins.
Protein PTM results from the enzymatic or nonenzymatic attachment of specific chemical groups to amino acid side chains. Such modifications occur either following protein translation or concomitant with translation. PTM influences both protein structure and physiological and cellular functions. Examples of enzymatic PTMs include phosphorylation, glycosylation, acetylation, methylation, sumoylation, palmitoylation, biotinylation, ubiquitination, nitration, chlorination, and oxidation/reduction. Nonenzymatic PTMs include glycation, nitrosylation, oxidation/reduction, acetylation, and succination. Some rare and unconventional PTMs, such as glypiation, neddylation, siderophorylation, AMPylation, and cholesteroylation, are also known to influence protein structure and function. Note that many of these modifications are not made in isolation. It is common for proteins to have several different types of modifications and that these modifications can differ depending on the tissue type and environmental circumstances present. The major PTMs in eukaryotes, their target amino acid residue(s), and the types of enzyme(s) or protein(s) involved are shown in Table 8.1.
Table 8.1 Common Protein Post-Translational Modifications, Their Target Amino Acid Residues, and the Enzyme(s) or Proteins Involved
![]()
Table from: Santos, A.L, and Lindner, A.B. (2017) Oxidative Medicine and Longevity, Article ID: 5716409
Methods to Detect Protein Posttranslational Modifications
Specific amino acid residues are subjected to PTMs depending on the chemistry of the reaction and the sequence specificity of the enzyme involved. Initially, the detection of PTMs was carried out by various analytical methods, such as radiolabeling of the proteins, thin-layer chromatography, column chromatography, and/or polyacrylamide gel electrophoresis. Other methods, such as protein sequencing by Edman degradation and Western blotting using protein-specific antibodies, have since been developed. Currently, antibody-based detection methods and mass spectrometry-based proteomic analysis are predominant methods used to detect and analyze PTMs. However, mass spectrometric methods are the only available tool to perform global or large-scale PTM analysis.
Antibody-based methods mostly rely on the availability of antibodies that can specifically recognize a modified amino acid residue within a protein or peptide. Such antibodies can be polyclonal or monoclonal and are developed against either the modified peptide/protein or against the modified amino acid. Moreover, antibody-based detection and quantification of PTMs on protein/peptide samples can be performed by two methods: chemiluminescence-based Western blotting and absorbance/fluorescence-based ELISA. However, the detection of PTMs depends entirely on the recognition site of the antibody used. If the antibody detects only the modified amino acid, additional analysis—for instance, protein/peptide isolation and sequencing—should be performed to detect the sequence context of the modification. However, if the antibody detects the PTM within a specific sequence context, the presence of PTM at other sites will remain undetected (ie the antibody will be specific for only that single modification).
Mass spectrometric detection of specific PTMs is based on mass changes. Depending on the type of modification, a specific change in mass of the modified amino acid or peptide occurs. Subsequently, the change in mass is detected by the mass spectrometer to identify the presence of a PTM in a peptide sample. Using tandem mass spectrometric methods, identification of the specific site of PTM can be achieved by subsequent fragmentation and sequencing of the relevant peptide. Yet, technical challenges hamper MS-based investigation of biologically important PTMs, such as ADP-ribosylation, one of the key signaling molecules that regulates DNA repair, a critical process in maintaining genome stability that is compromised in cancer and aging.
Data increasingly implicate PTMs not only during aging and/or under pathological conditions but also for the normal functioning of the cell. In turn, PTMs are increasingly studied for their role in health and disease. For example, the precise and accurate measurement of distinct PTM-containing moieties offers potential biomarker utility to aid early diagnosis, prognosis, monitoring response to therapy and decisions regarding inclusion in clinical trials as new medicines are developed. However, technical difficulties limit these studies, leaving many unanswered questions. The identification of unknown/unexpected PTMs by proteomic data reanalysis is an emerging subfield of proteomics recently boosted by the increased availability of raw data shared in public repositories. Notably, though, a sampling of the proteome in a given organism or cell provides only a snapshot of a highly dynamic process, confounding the analytical problem and ultimately arguing for time-resolved inventories. Thus, while many tools are currently available for the study of PTMs, new methods are needed to further advance the study of these modifications.
Examples of PTMs
Protein PTMs involve the covalent addition of some chemical group by enzymatic catalysis. Typically, an electrophilic fragment of a co-substrate is added to an electron-rich protein side chain, which acts as a nucleophile in the transfer. Common covalent protein PTMs include phosphorylation, acylation, methylation, sumoylation, ubiquitination, glycosylation, lipidation, oxidation and disulfide bond formation (either internal within a single protein or linking two protein/peptide chains together). Examples of common PTMs are provided in Figure 8.5.
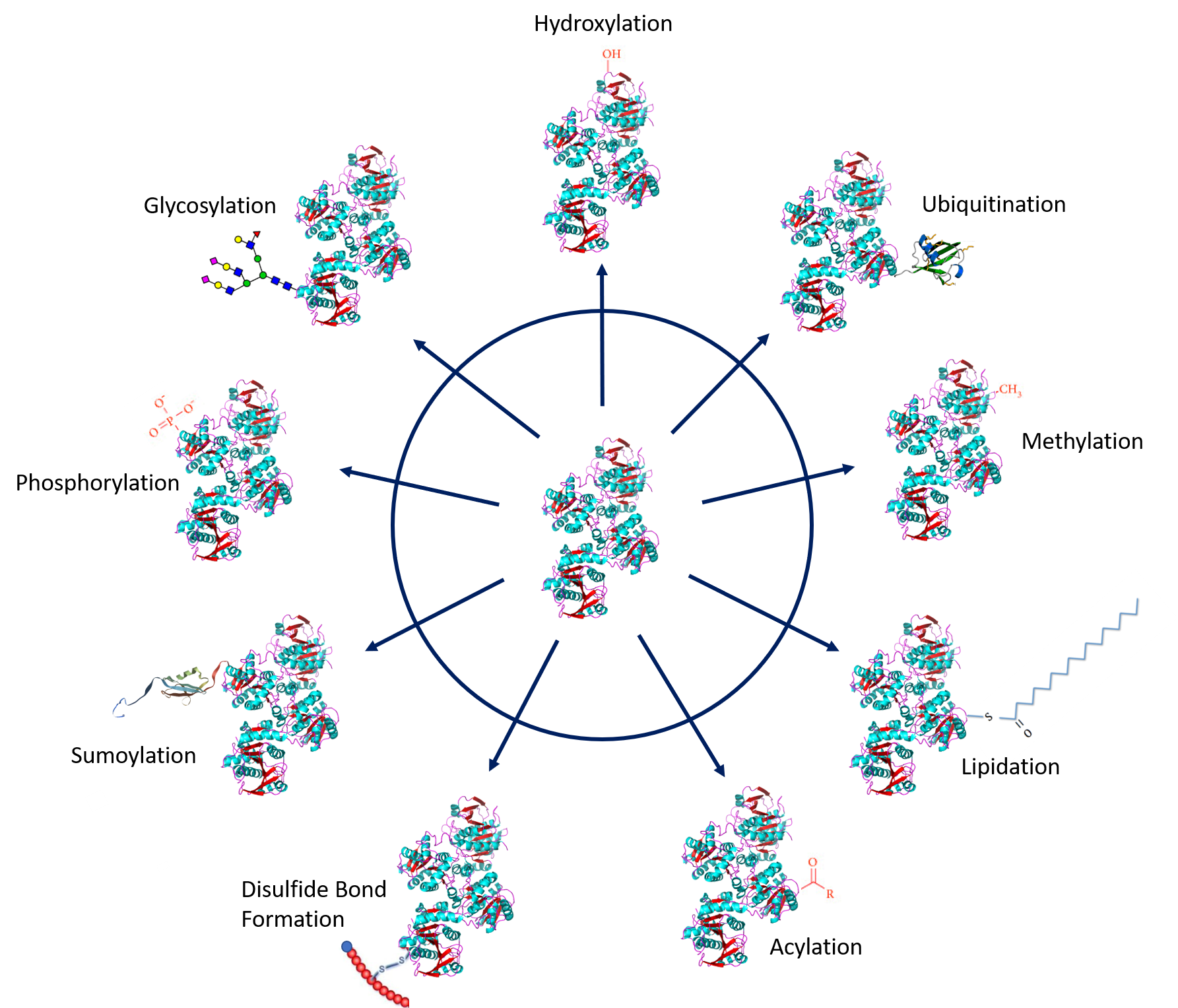
.jpg){kind=link}
{kind=link}
{kind=link}
Figure modified from: Santos, A.L, and Lindner, A.B. (2017) Oxidative Medicine and Longevity, Article ID: 5716409, Goettig, P. (2016) Int. J. Mol. Sci. 17(12), 1969, Rogerdodd, Jakob Suckale, Akinlade, A., et al (2014) Int. Archives of Med 7(50):28, WilsonNR, and Sivart13
{kind=link}
{kind=link}
back to the top
Protein Phosphorylation
One of the most common posttranslational modifications, protein phosphorylation, is the reversible addition of a phosphoryl group from adenosine triphosphate (ATP) to amino acid side chains such as serine, threonine, and tyrosine residues. This modification causes conformational changes that either (1) affect the catalytic activity to activate or inactivate the protein and/or cause the tendency of a protein to misfold and aggregate or (2) recruit other proteins to bind; both responses result in altered protein function and cell signaling. Phosphorylated proteins have critical and well-known functions in diverse cellular processes across eukaryotes, but phosphorylation also occurs in prokaryotic cells. In humans, about one-third of proteins are estimated to be substrates for phosphorylation. Indeed, phosphorylated proteins are now identified and characterized by high-throughput phosphoproteomics studies. The reversibility of protein phosphorylation is attributed to the actions of kinases and phosphatases, which phosphorylate and dephosphorylate substrates, respectively. The temporal and spatial balance of kinase and phosphatase concentrations within a cell mediates the size of its phosphoproteome.
Within a protein, phosphorylation can occur on several amino acids. Phosphorylation in eukaryotes most commonly occurs on serine residues, followed by threonine. Tyrosine phosphorylation is relatively rare but lies at the head of many protein phosphorylation signalling pathways (e.g. in tyrosine kinase-linked receptors, as shown in Figure 8.6). Histidine and aspartate phosphorylation occurs predominantly in prokaryotes as part of two-component signaling mechanism and in some cases in eukaryotes in some signal transduction pathways.
A notable example of proteins that are phosphorylated include Receptor Tyrosine Kinases. While tyrosine phosphorylation is found in relatively low abundance, it is well studied due to the ease of purification of phosphotyrosine using antibodies. Receptor tyrosine kinases are an important family of cell surface receptors involved in the transduction of extracellular signals such as hormones, growth factors, and cytokines. Binding of a ligand to a monomeric receptor tyrosine kinase stabilizes interactions between two monomers to form a dimer, after which the two bound receptors self-phosphorylate tyrosine residues on the inner side of the plasma membrane. Self-phosphorylation activates the receptor signaling pathway through enzymatic activity and facilitating interactions with adaptor proteins that recognize key phosphorylated tyrosine residues. Signaling pathways that utilize receptor tyrosine kinases are critical for a variety of cellular functions. For example, signaling through the epidermal growth factor receptor (EGFR), a receptor tyrosine kinase, is critical for the development of multiple organ systems including the skin, lung, heart, and brain. Furthermore, excessive signaling through the EGFR pathway can cause disease states and is found in many human cancers.
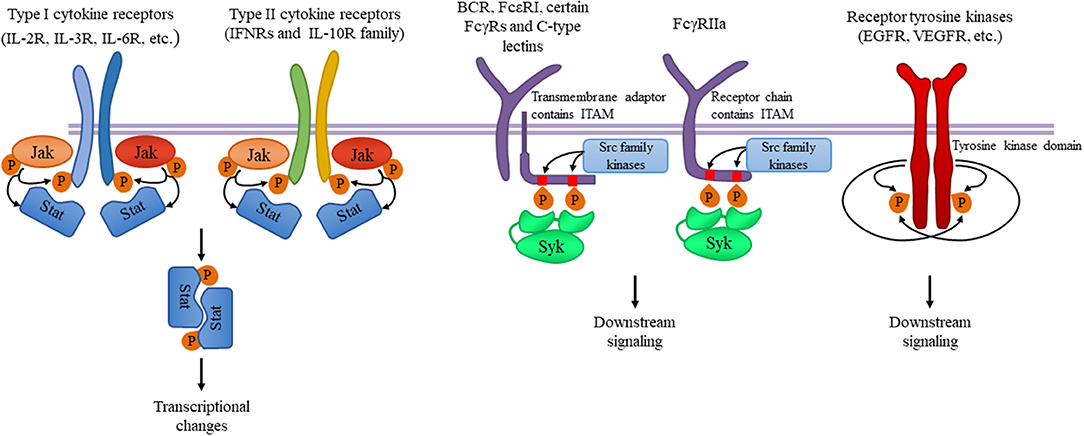
Figure 8.6. Tyrosine kinases and their signaling pathways. Type I and type II cytokine receptors utilize Janus kinases for the initiation of downstream signaling involved in the inflammatory responses. Src-family kinases and Syk are involved in several immune cell signaling pathways like immunoreceptor, integrin and C-type lectin signaling. Upon ligand binding, activation of Src-family kinases leads to the phosphorylation of tyrosine residues in immunoreceptor tyrosine-based activation motifs (ITAMs), that can be part of a transmembrane adaptor molecule like in case of B cell receptor (BCR), FcεRI, and certain FcγRs and C-type lectins, or of the receptor chain itself like in FcγRIIa in humans. Syk is recruited to the dually phosphorylated ITAMs and becomes activated resulting in the recruitment and activation of various further adapter proteins promoting downstream signaling. Receptor tyrosine kinases, for example EGFR and VEGFR have intrinsic tyrosine kinase activity leading to auto-and transphosphorylation of the receptor chains upon ligand binding. Recruitment of several adaptors and effector molecules through SH2 and phosphotyrosine binding domains mediate downstream signaling.
Figure from: Szilveszter, K.P., et al. (2019) Front. Immunol. Article ID: 10.3389(2019.01862)
Protein Acylation
The simplest form of acylation is protein N-Acetylation. N-acetylation is the reversible or irreversible transfer of an acetyl group to a nitrogen molecule through the actions of cleavage of methionine by methionine aminopeptidase (MAP) and the addition of an acetyl group from acetyl-CoA by N-acetyltransferase (NAT). Interestingly, 80–90% of eukaryotic proteins are acetylated, yet the underlying biological significance remains unclear. In the case of histone proteins (shown below in Figure 8.7), which make up chromatin, lysine acetylation regulates gene transcription, thereby affecting the cell’s transcriptome.
Histones are a highly basic family of proteins found in eukaryotic cell nuclei that pack and order the DNA into structural units called nucleosomes (Figure 8.7). They have multiple lysine and arginine residues that will interact with the negatively charged phosphate groups of the DNA backbone. The nucleosome core is formed of two H2A-H2B dimers and an H3-H4 tetramer, forming two nearly symmetrical halves by tertiary structure (C2 symmetry; one macromolecule is the mirror image of the other). The 4 ‘core’ histones (H2A, H2B, H3 and H4) are relatively similar in structure and are highly conserved through evolution, all featuring a ‘helix turn helix turn helix’ motif (a DNA-binding protein motif that recognize specific DNA sequence). They also share the feature of long ‘tails’ on one end of the amino acid structure – this being the location of post-translational modification, specifically N-acetylation.

Figure 8.7 Histone octomers form the backbone for chromosomal structure. Acetylation and methylation affect histone-DNA interactions. DNA methylation increases histone-DNA affinity and blocks transcriptional activation. Acetylation of the histone tails disrupts histone-DNA interactions and facilitates gene expression.
Figure from: Richard Wheeler
{kind=link}
Histone acetylation typically results in transcriptional activation; deacetylation typically results in transcriptional suppression. Acetylation occurs via histone acetyltransferases (HATs) and is reversible via the action of histone deacetylases (HDACs). One group of histone deacetylases are the sirtuins (silent information regulator), which maintain gene silencing via hypoacetylation. Sirtuins have been reported to aid in maintaining genomic stability.
Although first described in histones, acetylation is also observed in cytoplasmic proteins. Acetylated proteins can also be modulated by the cross-talk with other posttranslational modifications, including phosphorylation, ubiquitination, and methylation. Therefore, acetylation may contribute to cell biology beyond transcriptional regulation.
back to the top
Protein Glycosylation
Protein glycosylation involves the addition of a diverse set of sugar moieties to the protein core. Glycosylation has significant implications for protein folding, conformation, distribution, stability, and activity. Glycosylated proteins can have additions of simple monosaccharides. For example, many nuclear transcription factors are modified in this way. Alternatively, some proteins are modified with highly complex branched polysaccharides, such as those seen on cell surface protein receptors, as shown in Figure 8.8.
More than half of all mammalian proteins are believed to be glycosylated. However, glycoprotein functions, at both molecular and cellular levels, remain unclear. While proteins exhibit improved stability and trafficking after glycosylation in vivo, glycan structures can alter protein functions or activities. These structures often result from the activities of glycan-processing enzymes working within a cell at any given time. However, the structures are sometimes protein-specific, depending on protein trafficking properties and interactions with other cellular factors.
There are three types of protein glycosylation in higher eukaryotes: N-linked, O-linked, and C-linked. These types reflect their glycosidic linkages to amino acid side chains. In N-linked glycosylation, β-N-acetylglucosamine (GlcNAc) is attached through an amide linkage to the side chain of Asn within a consensus sequence of AsnXaaSer/Thr (Figure 8.8). N-linked glycans have multiple functions. While they act as ligands for glycan-binding proteins in cell-cell communication, they also can regulate glycoprotein aggregation in the plasma membrane and affect the half-life of antibodies, cytokines, and hormones in serum.
O-linked glycosylation in higher eukaryotes occurs through several different mechanisms. The most abundant type of O-linked glycosylation is the mucin-type, involved attachment of an α-N-acetylgalactosamine (GalNAc) to the hydroxyl group of Ser/Thr side chains. Mucins are a family of high molecular weight, heavily glycosylated proteins (glycoconjugates) produced by epithelial tissues in most animals. A key characteristic of mucin proteins is their ability to form gels; therefore they are a key component in most gel-like secretions, serving functions from lubrication to cell signalling to forming chemical barriers. Aberrant expression of mucin-type O-linked glycans occurs in cancer cells and may provide targets for anticancer vaccines.
O-linked glycosylation occurring with the addition of α-O-mannose is the only form of O-linked glycosylation in yeast, but also occurs in the brains of higher eukaryotes. Higher eukaryotes also have an α-O-fucose modification of Ser/Thr residues. This type of glycosylation modulates signaling pathways during eukaryotic development. Another modification, β-O-galactosylation, may contribute to rheumatoid arthritis.
Finally, C-linked glycosylation involves the addition of α-mannose (Man) to the 2-position of the indole side chain of tryptophan residues. While first identified on ribonuclease 2, it also occurs on other proteins, including some of the mucin proteins, thrombospondin, and the Ebola virus soluble glycoprotein.
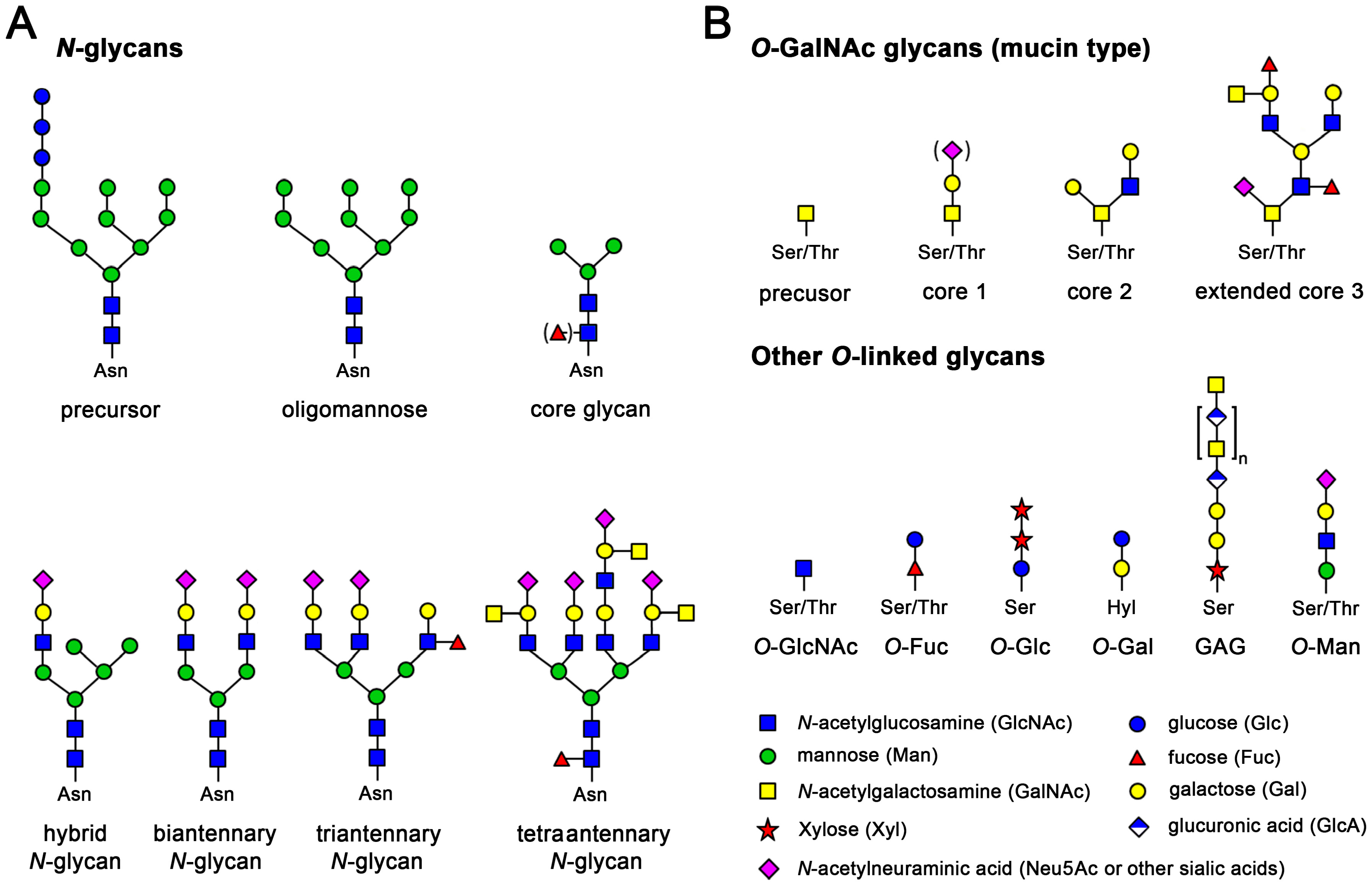
Figure 8.8. Examples of the Most Common N-linked and O-linked Glycosylation Patterns. (A) N-glycosylation of asparagine in protein regions with the consensus sequence Asn-Xaa-Ser/Thr. N-glycans are generated by trimming and extending the common precursor GlcNAc2Man9Glu3. Small core glycans are mostly intermediates in mammalian glycan synthesis, but often occur in more primitive eukaryotes and insects, as used for recombinant expression. Mammalian N-glycans exhibit an enormous diversity, due to many possible combinations of branching sugars; (B) O-glycosylation at Ser and Thr is found in all kingdoms of life. There is no distinct consensus sequence, but proline-rich regions are favored, e.g., a typical O-glycan site would be Pro-Ser/Thr-Xaa-Yaa-Pro. A very common mammalian O-glycan is the mucin-type that starts with GalNAc and is extended by galactose and sialic acids or GlcNAc, with eight different cores known. In addition, the O-xylose linked, non-branched glucosamine glycans (GAG) or proteoglycans are a large and diverse glycan family. The displayed chondroitin can be phosphorylated and heavily sulfated, comprising up to fifty disaccharide units. O-GlcNAc glycans occur inside cells, even in the nucleus, while O-galactosylation is found at hydroxylysine residues (Hyl) of collagens
Figure from: Goettig, P. (2016) Int. J. Mol. Sci. 17(12), 1969
Protein Ubiquitination and Sumoylation
Ubiquitination is the addition of an 8 kDa polypeptide, called ubiquitin, to lysine residues of target proteins via the C-terminal glycine residue of ubiquitin. The addition of one ubiquitin causes the formation of a ubiquitin polymer. If the protein target is monoubiquinated, this can alter the activity of the target protein. For example monoubiquitination of histone proteins promotes the release the DNA enabling transcription. However, if multiple ubiquitins are added to the substrate forming a polyubiquitinated protein, this typically targets the protein for the 26S proteasome where it will be degraded. Details of this degradation pathway are described in section 8.5.
Similarly, protein sumoylation is a reversible posttranslational modification whereby a small ubiquitin-like modifier (SUMO) protein is covalently attached to target. Like ubiquitin, SUMO is conjugated to the lysine side chains of target proteins, and it is removed by SUMO-specific isopeptidases. Over the last few decades, it has been well established that sumoylation controls many aspects of nuclear function. However, recent research has started to unveil a determinant role of protein sumoylation in many extranuclear neuronal processes and potentially in a wide range of neuropathological conditions.


Figure 8.9 Structures of the Ubiquitin and SUMO Proteins. Upper structure schematic of ubiquitin protein, highlighting the secondary structure. α-helices are colored in blue and β-strands in green. The sidechains of the 7 lysine residues are indicated by orange sticks. The two best-characterised attachment points for further ubiquitin molecules in polyubiquitin chain formation (lysines 48 & 63) are labelled. Lower structure schematic of human SUMO1 protein made with iMol and based on PDB file 1A5R, an NMR structure; the backbone of the protein is represented as a ribbon, highlighting secondary structure; N-terminus in blue, C-terminus in red
Upper Figure is from: Rogerdodd and the lower figure is from Jakob Suckale
back to the top
Protein Oxidation
The reaction of proteins with a variety of free radicals and reactive oxygen species (ROS) leads to oxidative protein modifications such as formation of protein hydroperoxides, hydroxylation of aromatic groups and aliphatic amino acid side chains, oxidation of sulfhydryl groups, oxidation of methionine residues, conversion of some amino acid residues into carbonyl groups, cleavage of the polypeptide chain, and formation of cross-linking bonds. Aromatic and sulfur-containing amino acid residues are particularly susceptible to oxidative modification.
Unless repaired or removed from cells, oxidized proteins are often toxic and can impair cellular viability, since oxidatively modified proteins can form large aggregates. Oxidatively damaged proteins undergo selective proteolysis, primarily by the 26S proteasome in an ubiquitin- and ATP-independent way. Ultimately, upon extensive protein oxidation, these aggregates can become progressively resistant to proteolytic digestion and actually bind the 20S proteasome and irreversibly inhibit its activity.
Protein carbonylation is defined as an irreversible posttranslational modification (PTM) whereby a reactive carbonyl moiety, such as an aldehyde, ketone, or lactam, is introduced into a protein. The first identified source of protein-bound carbonyls was metal-catalyzed oxidation (MCO). MCO results from the Fenton reaction when transition metal ions are reduced in the presence of hydrogen peroxide, generating the highly reactive hydroxyl radicals in the process. These hydroxyl radicals can oxidize amino acid side chains or cleave the protein backbone, leading to numerous modifications including reactive carbonyls. For example, oxidation of proline and arginine results in the production of glutamic semialdehyde, while lysine is oxidized to aminoadipic semialdehyde and threonine to 2-amino-3-ketobutyric acid. Direct oxidation of other amino acid residues can also lead to protein-bound carbonyls. Tryptophan oxidation by ROS produces at least seven oxidation products. Among them are kynurenine and N-formyl kynurenine, as well as their hydroxylated analogs, which contain aldehyde or keto groups formed by oxidative cleavage of the indole ring.
Another important source of protein-bound carbonyls is reactive lipid peroxidation products, which are produced during oxidation of polyunsaturated fatty acids. Protein carbonylation can also occur via glycoxidation. Reactive α-carbonyls formed during glycoxidation, such as glyoxal, methylglyoxal, and 3-deoxyglucosone, can then modify the basic residues Lys and Arg to generate, for example, pyrralines and imidazolones. Glycation (i.e., the reaction of reducing sugars such as glucose or fructose with the side chains of lysine and arginine residues) forms Amadori and/or Hynes products. These glycated residues can be further decomposed by ROS into advanced glycation end products (AGE) carrying carbonylated moieties that can also contribute for protein carbonylation.
Some oxidative modifications are made enzymatically and have key regulatory or structural functions within the modified proteins. For example, proline can be converted to hydroxyproline and lysine to hydroxylysine, as shown in figure 8.10. 4-Hydroxyproline makes up about 13.5% of the residues within the mammalian collagen family of proteins. Recall that collagen is the main protein of the connective tissue and represents about one-fourth of the total protein content in many animals. Hydroxyproline contributes to the stability of the triple helix and also aids in cross-linking between collagen fibers to form larger macromolecular complexes.

Figure 8.10 Hydroxyproline and Hydroxylysine. Schematic drawing to show protein hydroxylation occurring at ( a ) proline and ( b ) lysine to form hydroxyproline (HyP) and hydroxylysine (HyL), respectively.
Figure from Xu, Y., et. al. (2014) Int J Mol Sci 15(5):7594-7610.
Protein Methylation
Alkyl substituents are also a common post translational modification. The introduction of such alkyl groups results in the alteration of the hydrophobicity of the modified protein. The most common type of protein alkylation is protein methylation, which is mediated by methyltransferase enzymes. One-carbon methyl groups are added to nitrogen or oxygen (N- and O-methylation, resp.) on amino acid side chains, increasing protein hydrophobicity or neutralizing a negative charge when bound to carboxylic acids. While N-methylation is typically irreversible, O-methylation is potentially reversible. Methylation occurs so often that its primary methyl donor, S-adenosyl methionine (SAM), is one of the most-used enzymatic substrates after ATP (Figure 8.11).

Figure 8.11 S-Adenosylmethionine (SAM). The methyl group attached to the sulfur residue is donated to protein targets during protein methylation.
Figure from: Alsosaid 1987
{kind=link}
A common theme with methylated proteins, as is also the case with phosphorylated proteins, is the role this modification plays in the regulation of protein-protein interactions. For instance, the arginine methylation of proteins can either inhibit or promote protein-protein interactions depending on the type of methylation. Protein methylation is also a common modification found in the histone protiens. The transfer of methyl groups from S-adenosyl methionine to histones is catalyzed by enzymes known as histone methyltransferases. The N-terminal tails of histones H3 and H4 receive methyl groups on specific lysines. Methylation then determines if gene transcription is activated or repressed, thus leading to different biological outcomes.
Histone methylation was traditionally thought to be irreversible. However, histone demethylases demonstrate the reversibility of this PTM. Indeed, dynamic modifications to chromatin were imposed by an ability or inability to maintain equilibrium in the opposing effects of methylases and demethylases. The simultaneous removal of one histone methylation and an addition of another can enable transcriptional fine tuning.
In addition to methylation, histone acetylation, deacetylation, and mono-ubiquitination are also essential parts of gene regulation (Figure 8.12). Acetylation removes the positive charge on the histones, thereby decreasing the interaction of the N termini of histones with the negatively charged phosphate groups of DNA. As a consequence, the condensed chromatin is transformed into a more relaxed structure that is associated with greater levels of gene transcription.
Nonhistone proteins also exhibit methylation as a common PTM that regulates signal transduction pathways. Furthermore, methylation works in concert with other types of PTMs, as well as with histone and nonhistone proteins, to exert influence on not only chromatin remodeling but also gene transcription, protein synthesis, and DNA repair.
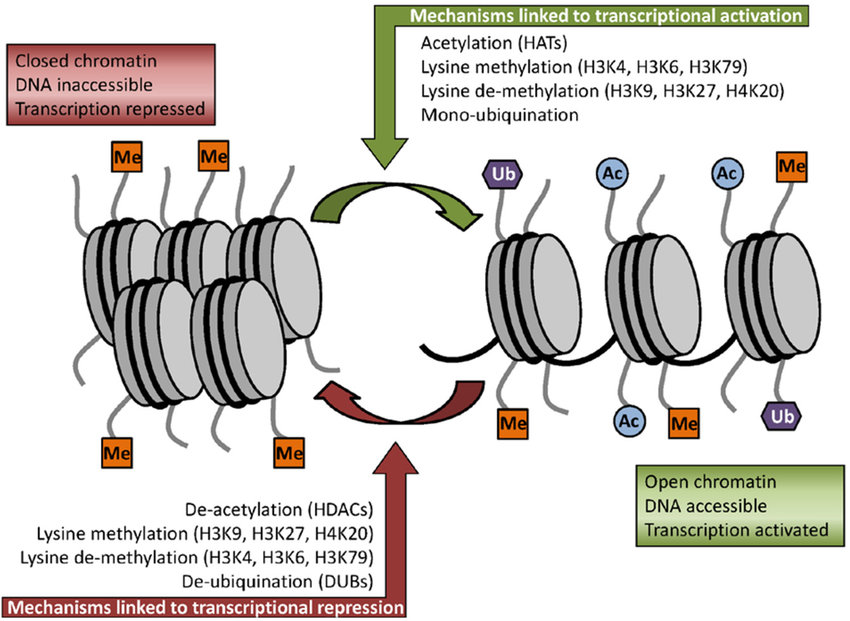
Figure 8.12 Some of the key histone modifications influencing gene expression (Me: methylation, Ub: ubiquination, Ac: acetylation).
Figure from: Ullah, M.F. (2015) Medicines 2:141-156.
back to the top
8.3 Allosteric Regulation
Allosteric regulation fine-tunes most biological processes, including signal transduction, enzyme activity, metabolism and transport. Allostery, an intrinsic property of a protein, is referred to as the regulation of activity at one site (also known as an orthosteric site) in a protein by a topographically and spatially distant site; the latter is designated as an allosteric site. Allosteric regulation occurs through binding of a modulator (e.g., small molecule or protein) at an allosteric site to engender a conformational change that affects function at the orthosteric site. This effect may cause the re-distribution of the conformational ensemble by either stabilizing an active conformation (allosteric activation) or destabilizing an inactive conformation (allosteric inhibition) in response to allosteric perturbations (Figure 8.13). Traditionally, the repertoire of allostery was primarily confined to determining the allosteric effects or mechanisms in individual multi-subunit or monomer proteins by conformational transitions. Recently, increasing evidence has indicated that allosteric signals can propagate across several or numerous proteins to sculpt allosteric networks.
Allosteric regulation is particularly important in the cell’s ability to adjust enzyme activity based on the surrounding environmental conditions. Feedback control loops, such as feedback inhibition from downstream products or feedforward from upstream substrates are common allosteric regulatory mechanisms found in nature. Another example of allostery includes oxygen binding to one of the subunits of hemoglobin that prompts cooperative binding to other subunits.

Figure 8.13 Allosteric Regulation of an Enzyme. Effectors that enhance the protein’s activity are referred to as allosteric activators, whereas those that decrease the protein’s activity are called allosteric inhibitors.
Figure by: Tenthkrige
{kind=link}
Many allosteric effects can be explained by the concerted MWC model put forth by Monod, Wyman, and Changeux, or by the sequential model described by Koshland, Nemethy, and Filmer. Both postulate that protein subunits exist in one of two conformations, tensed (T) or relaxed (R), and that relaxed subunits bind substrate more readily than those in the tense state. The two models differ most in their assumptions about subunit interaction and the preexistence of both states.
Concerted model
The concerted model of allostery, also referred to as the symmetry model or MWC model, postulates that enzyme subunits are connected in such a way that a conformational change in one subunit is necessarily conferred to all other subunits. Thus, all subunits must exist in the same conformation. The model further holds that, in the absence of any ligand (substrate or otherwise), the equilibrium favors one of the conformational states, T or R. The equilibrium can be shifted to the R or T state through the binding of one ligand (the allosteric effector or ligand) to a site that is different from the active site (the allosteric site).
Sequential model
The sequential model of allosteric regulation holds that subunits are not connected in such a way that a conformational change in one induces a similar change in the others. Thus, all enzyme subunits do not necessitate the same conformation. Moreover, the sequential model dictates that molecules of a substrate bind via an induced fit protocol. While such an induced fit converts a subunit from the tensed state to relaxed state, it does not propagate the conformational change to adjacent subunits. Instead, substrate-binding at one subunit only slightly alters the structure of other subunits so that their binding sites are more receptive to substrate. To summarize:
- subunits need not exist in the same conformation
- molecules of substrate bind via induced-fit protocol
- conformational changes are not propagated to all subunits
back to the top
8.4 Zymogen Activation
A zymogen also called a proenzyme, is an inactive precursor of an enzyme. A zymogen requires a biochemical change (such as a hydrolysis reaction revealing the active site, or changing the configuration to reveal the active site) for it to become an active enzyme.
An example of enzymes that are initially synthesized as zymogens are the protease enzymes secreted by the pancreas. The pancreas secretes zymogens to help prevent the enzymes from inappropriately digesting proteins in the pancreatic cells in which they are synthesised. Enzymes like Trypsin are synthesized as proenzymes. For typsin, trypsinogen is an inactive precursor that is translated in the rough endoplasmic reticulum and transported to the Golgi apparatus for sorting. Trypsinogen is always co-synthesized and packed with a pancreatic secretory trypsin inhibitor (PSTI) that inhibits its premature activation. Thus, there are two mechanisms in place to maintain the inactivity of the protease within the pancreas: (1) synthesis of the zymogen or proenzyme form, and (2) co-expression of a trypsin inhibitor protein that will bind and inhibit any prematurely cleaved trypsin until it has reached the small intestine (Figure 8.14).

Figure 8.14 Trypsinogen and Trypsin. On the left, the precursor zymogen, Trypsinogen, is shown. The portion of the inactivating prepeptide that can be visualized during crystalization is shown in green. On the right, Trypsin is shown. The new conformation of the protein without the prepeptide is indicated by the change in the pink amino acid residues that are tucked into the protein structure in the active conformation. Within zymogen granules, small amounts of Trypsin are formed by the inappropriate cleavage of Trypsinogen. Thus, a small Trypsin Inhibitor, shown in red, is coexpressed by pancreatic cells and binds with any of the activated Trypsin, keeping it in the inactive state until it is secreted into the small intestine.
Figure from: Goodsell, D. (2012) Molecule of the Month, Protein Database
During packaging within the Golgi system, the trypsinogen and other digestive enzymes condense into core particles and are packed in zymogen granules. The condensed enzymes are stable and minimal activation happens within the zymogen granules. Once the pancreatic cells receive secretory stimulus, these zymogen granules are released in to the lumen of pancreatic duct, which carries the digestive enzymes into the duodenum. Once in duodenum, enteropeptidase activates trypsinogen by removing 7-10 amino acid from N-terminal region known as trypsinogen activation peptide (TAP) (Figure 8.15). Removal of TAP induces conformational change resulting in active trypsin. TAP is immunologically distinct from the same sequence within trypsinogen, thereby allowing detection of trypsinogen activation in situ.
Once activated, trypsin will cleave and activate other zymogen proteases and lipases present in the duodenum. These include the activation of elastase, chymotrypsin, carboxypeptidase, and lipase (Figure 8.15) Zymogens are also found in other cellular processes as well. For example, intracellular proteases known as caspases, are activated in a similar manner during the process of cellular apoptosis or programmed cell death. The process of blood clotting also involves the activation of zymogens.

Figure 8.15 Zymogen Activation Cascade in the Small Intestine.
Figure by Joyjiang
{kind=link}
back to the top
8.5 Intracellular Protein Degradation
The 26S proteasome is the central element of proteostasis regulation in eukaryotic cells. It is required for the degradation of protein factors in multiple cellular pathways and it plays a fundamental role in cell stability. The main aspects of proteasome-mediated protein degradation have been largely described during the last three decades with the use of intense cellular, molecular, structural and chemical biology research and tool development. The 26S proteasome has a structural configuration that confines the proteolytic active sites in a location unreachable for native and functional proteins, thus preventing uncontrolled degradation. The proteolytic active sites are found in the interior of a barrel-shaped core particle (CP or 20S). The entrances of the tunnel, placed at the distal ends of the barrel, are commonly occupied by the regulatory particle (RP or 19S), a sophisticated protein assembly that acts as a substrate processing machine. the regulatory particle has the important role of receiving, deubiquitinating, unfolding and translocating substrates to the CP and it adopts different configurations depending on the activity states they exhibit. This process typically requires ATP hydrolysis. Moreover, conformationally distinct proteasomes may show different subcellular distributions depending on functional requirements in each cellular type and environmental situations. Proteasomes are distributed throughout the cell, detected in the cytoplasm and in the nucleus, and they can localize to hotspots in distinct intracellular regions or specific sites with high protein metabolism or with specific protein degradation requirements.

Figure 8.16 Structure of a Yeast Proteosome. The core particle of the 26S proteosome is shown here in yellow and red. Within the core, three types of proteases, shown in different shades of red, are present and each have specific affinity for specific peptide sequences. This ensures maximal breakdown of proteins that are targeted for degradation. ATP binds within the magenta portions of the proteosomes and provides the needed energy to unfold proteins targeted for degradation and prepares them for cleavage in the peptidase-containing core. The regulatory subunits, shown in blue are responsible for the proteosome selectivity, ensuring that only proteins targeted for degradation are processed.
Figure from: Goodsell, D. (2012) Molecule of the Month, Protein Database
Proteins are normally targeted to the proteasome by means of ubiquitin labels attached covalently to a lysine residue, usually in a chained form that produces a polyubiquinated protein. The process of protein polyubiquitination is carried out by a highly specialized and diverse enzymatic system, which
includes families of ubiquitin activating enzymes (E1), ubiquitin conjugating enzymes (E2) and ubiquitin ligases (E3).
The covalent attachment of ubiquitin to specific target proteins is mainly accomplished by stepwise enzymatic cascade reactions, and ubiquitin is attached to the substrates via the concerted action of ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin ligase (E3). The attachment of ubiquitin or ubiquitin chains to the substrate is a successive process (Figure 8.17). First, the C-terminal carboxylic acid is activated by adenylation using a molecule of ATP forming an adenylate (AMP-) intermediate. The adenylate acts as a good leaving group during the next reaction where an E1-ubiquitin thioester bond is formed between the C-terminal Gly carboxyl group of ubiquitin and the active site Cys of the E1 enzyme. AMP leaves the active site at this point. Note that ATP is used in many reactions to activate carboxylic acid functional groups through the formation of an adenylate intermediate and that this will be seen as a theme in many different types of reactions throughout this textbook. Once the ubiquitin is docked as a thioester on E1, it can be transferred to a Cys residue of the E2 enzyme to form an E2-ubiquitin thioester-linked intermediate. This enzymatic reaction is known as a transesterification.
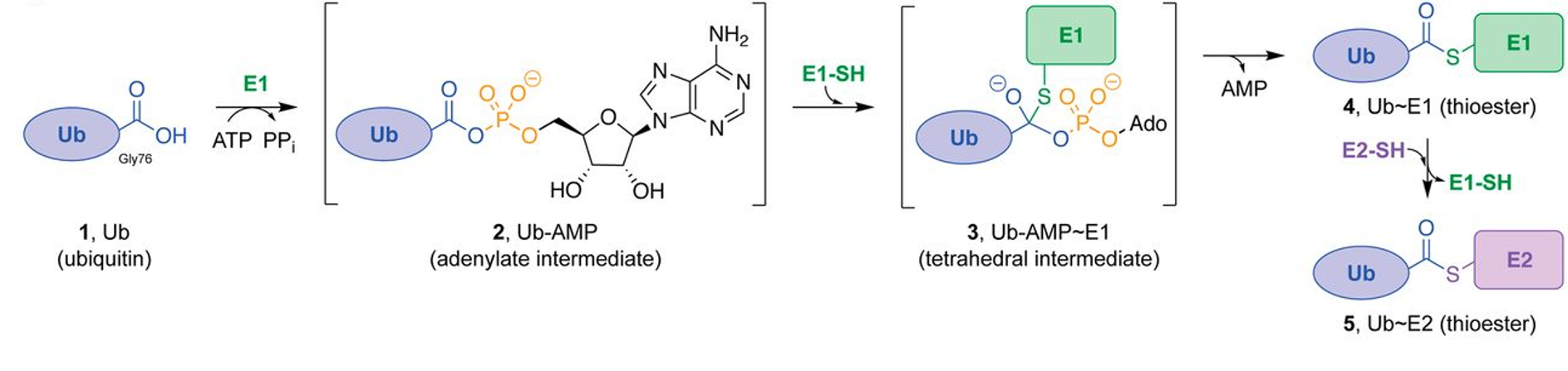
Figure 8.17 Enzymatic Activity of the E1 Ubiquitin Activating Enzyme. Ub E1s catalyze adenylation of the Ub C-terminal glycine-76 (1 → 2) using a molecule of ATP, thioesterification with an E1 catalytic cysteine ensues (2 → 3 → 4), and transthioesterification to an E2 catalytic cysteine (4 → 5) completes the reaction.
Figure from: Hann, Z.S., et. al. (2019) PNAS 116(31)15475-15484.
Eventually the E2 transfers the ubiquitin to the substrate protein by E3. Ubiquitin is conjugated to the target protein through an isopeptide bond between its C-terminal glycine (Gly76) and the ε-amino group of a lysine residue. There are three typical ways of linking the ubiquitin with the substrate (Figure 8.18). The first is called mono-ubiquitination, which refers to the modification of one site of the modification of a substrate by a single ubiquitin molecule. The second is multi-mono-ubiquitination, which means adding several ubiquitin molecules repetitively to distinct sites (multi-mono-ubiquitination). The third is called polyubiquitination (including linear polyubiquitination and branched polyubiquitination), in which ubiquitin molecules are added to the same site (polyubiquitination, including linear polyubiquitination and branched polyubiquitination) of a substrate. In the second and third ways of linking, the previously attached ubiquitin serves as the “acceptor” of subsequently added ubiquitin. Of course, polyubiquitin chains linked by the same Lys are homogeneous, while those linked at different Lys are heterogeneous or mixed ones.
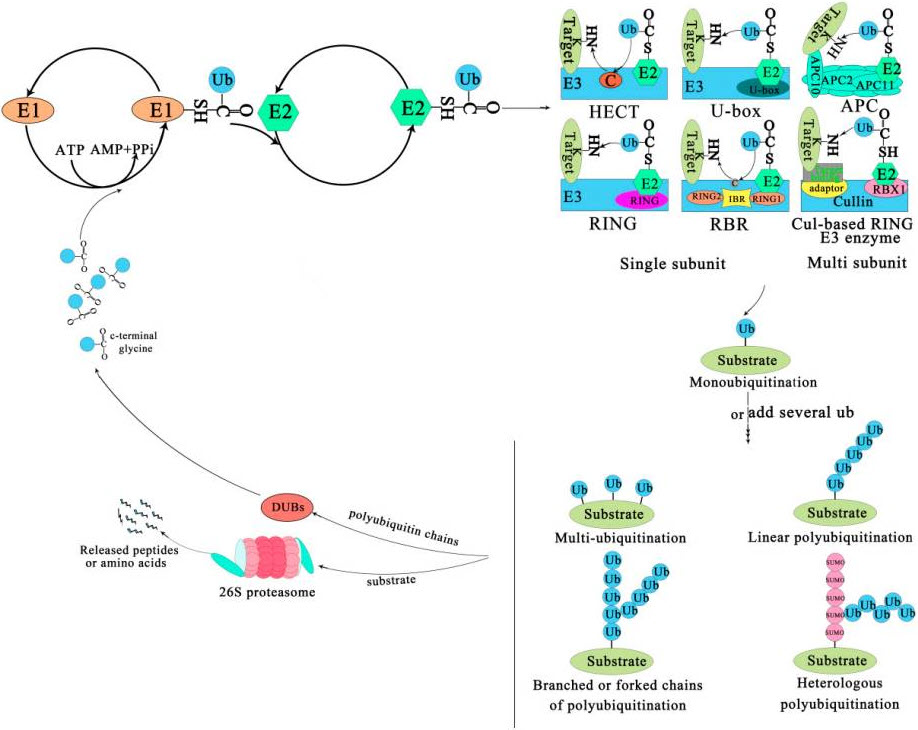
Figure 8.18 The Ubiquitin-Proteasome System. The process of ubiquitination from activation to attachment to the substrate is catalyzed by three major enzymes. The substrates labeled by ubiquitin are degraded by the 26S proteasome or play a non-degradative role in other processes. Abbreviations: APC, Anaphase-promoting complex; DUBs, Deubiquitinating enzymes; E1, Ubiquitin-activating enzyme; E2, Ubiquitin-conjugating enzyme; E3, Ubiquitin-ligase enzyme; Cul-based, Cullin-RING box1-Ligase; HECT, Homology to E6-AP C Terminus; Ub, Ubiqitin; SUMO, Small ubiquitin-related modifier; RBX1, RING-Box 1; RING, Really interesting new gene; RBR, RING1-IBR(cysteine/histidine rich region)-RING2.
Figure from: Liu, W., et.al. (2020) Int J Mol Sci 21(8):2894
Subsequently, the substrate complex tagged by the ubiquitin is either degraded by the 26S proteasome or executes nonproteolytic functions, such as the regulation of gene expression, cellular trafficking, or other biological function. In most cases, polyubiquitinated proteins are recognized and degraded by the 26S proteasome, and the ubiquitin or ubiquitin chain is hydrolyzed and freed by deubiquitinating enzymes (DUBs) for reuse in further conjugation cycles after being removed from the substrate protein (Figure 8.18).
The family of E3 enzymes is large and diverse. It is estimated that there are 600-700 E3 enzymes in humans, representing approximately 5% of the human genome. Thus, E3 enzymes can be very substrate specific, leading to the specialized degradation of a small subset of proteins within the cell. E2 enzymes are the intersection between E1 and E3 enzymes and help to determine the ubiquitination of specific target proteins by interacting with different types of E3 enzymes.
back to the top
8.6 References
- Shen, Q., Wang, G., Li, S., Liu, X., Lu, S., Chen, Z., Song, K., Yan, J., Geng, L, Huang, Z., Huang, W., Chen, G., and Zhang, J. (2016) ASDv3.0: Unraveling allosteric regulation with structural mechanisms and biological networks. Nucleic Acids Research 44(D1):D527-D535. Available at: https://academic.oup.com/nar/article/44/D1/D527/2503129
- Wikipedia contributors. (2020, January 19). Isozyme. In Wikipedia, The Free Encyclopedia. Retrieved 03:42, May 19, 2020, from https://en.wikipedia.org/w/index.php?title=Isozyme&oldid=936548836
- Wikipedia contributors. (2020, April 30). COX-2 inhibitor. In Wikipedia, The Free Encyclopedia. Retrieved 07:00, May 22, 2020, from https://en.wikipedia.org/w/index.php?title=COX-2_inhibitor&oldid=954080651
- Clarkson, C.W. (2018) Major Side Effects of NSAIDs and COX-2 Selective Inhibitors. TUSOM Pharmwiki. Available at: http://tmedweb.tulane.edu/pharmwiki/doku.php/nsaid_side_effects?do=
- Santos, A.L. and Lindner, A.B. (2017) Protein Porsttranslational Modifications: Roles in Aging and Age-Related Disease. Oxidative Medicine and Cellular Longevity, Article ID: 5716409. Available at: https://www.hindawi.com/journals/omcl/2017/5716409/#copyright
- Wikipedia contributors. (2020, May 12). Protein phosphorylation. In Wikipedia, The Free Encyclopedia. Retrieved 17:38, May 25, 2020, from https://en.wikipedia.org/w/index.php?title=Protein_phosphorylation&oldid=956228409
- Szylveszter, K.P., Németh, T., and Mócsai, A. (2019) Tyrosine Kinases in Autoimmune and Inflammatory Skin Diseases. Front. Immunol. 10.3389(2019.01862). Available at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.01862/full
- Wikipedia contributors. (2020, May 7). Histone. In Wikipedia, The Free Encyclopedia. Retrieved 21:12, May 25, 2020, from https://en.wikipedia.org/w/index.php?title=Histone&oldid=955458038
- Wikipedia contributors. (2020, May 13). Mucin. In Wikipedia, The Free Encyclopedia. Retrieved 01:10, June 7, 2020, from https://en.wikipedia.org/w/index.php?title=Mucin&oldid=956387296
- Wikipedia contributors. (2020, March 15). Allosteric regulation. In Wikipedia, The Free Encyclopedia. Retrieved 04:07, June 7, 2020, from https://en.wikipedia.org/w/index.php?title=Allosteric_regulation&oldid=945637073
- Dixit, Ajay. Dawra, Rajinder K. Dudeja, Vikas. Saluja, Ashok K. (2016). Role of trypsinogen activation in genesis of pancreatitis.
Pancreapedia: Exocrine Pancreas Knowledge Base, DOI: 10.3998/panc.2016.25 - Coll-Martinez, B., and Crosas, B. (2019) How the 26S Proteasome Degrades Ubiquitinated Proteins in teh Cell. Biomolecules 9(9):395. Retrieved from: https://www.mdpi.com/2218-273X/9/9/395
- Liu, W., Tang, X., Qi, X., Ghimire, S., Ma, R., Li, S., Zhang, N., and Si H. (2020) The Ubiquitin Conjugating Enzyme: An Important Ubiquitin Transfer Platform in Ubiquitin-Proteosome System. Int J Mol Sci 21(8):2894. Retrieved from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7215765/