Home » Student Resources » Online Chemistry Textbooks » CH450 and CH451: Biochemistry - Defining Life at the Molecular Level » Chapter 7: Catalytic Mechanisms of Enzymes
MenuCH450 and CH451: Biochemistry - Defining Life at the Molecular Level
Chapter 7: Catalytic Mechanisms of Enzymes
7.1 Concept Review for Enzyme Reactions
-
Definition of an Enzyme
-
Homolytic vs Heterolytic Bond Cleavage
-
Nucleophiles and Electrophiles
-
Electron Pushing
-
Acid Dissociation Constants, pKa
-
Primary Enzyme Classifications
7.2 Overview of Catalytic Mechanisms
-
Covalent Catalysis
-
Acid-Base Catalysis
-
Electrostatic Catalysis
-
Desolvation
-
Catalysis by Approximation
-
Coenzyme Catalysis
-
Strain Distortion
7.3 Examples of Reaction Mechanisms
7.4 References
7.1 Concept Review for Enzyme Reactions
In this section, we will review some fundamentals of organic and biological chemistry that are helpful in understanding enzyme reaction mechanisms.
Definition of an Enzyme
Recall from Chapter 6, that enzymes are biological catalysts that reduce the activation energy required for a reaction to proceed in the forward direction (Figure 7.1). They facilitate the formation of the transition state species within the reaction and speed up the rate of the reaction by a million-fold in comparison to non catalyzed reactions. Note that enzymes do NOT alter the ΔG of the reaction and do NOT have any affect on the spontaneity or equilibrium position of the reaction. Enzymes, like other catalysts, are also not used up during the reaction. Enzymes have high substrate specificity, and can even show regiospecificity that leads to the generation of stereospecific products.
Figure 7.1 Effect of an enzyme on reducint the activation energy required to start a reaction where (a) is uncatalyzed and (b) is an enzyme-catalyzed reaction.
Figure from Peter K. Robinson
Homolytic vs Heterolytic Bond Cleavage
Bond cleavage, or scission, is the splitting of chemical bonds. This can be generally referred to as dissociation when a molecule is cleaved into two or more fragments. In general, there are two classifications for bond cleavage: homolytic and heterolytic, depending on the nature of the process (Figure 7.2).
In heterolytic cleavage, or heterolysis, the bond breaks in such a fashion that the originally-shared pair of electrons remain with one of the fragments. Thus, a fragment gains an electron, having both bonding electrons, while the other fragment loses an electron.[4] This process is also known as ionic fission.
In homolytic cleavage, or homolysis, the two electrons in a cleaved covalent bond are divided equally between the products. This process is also known as homolytic fission or radical fission.
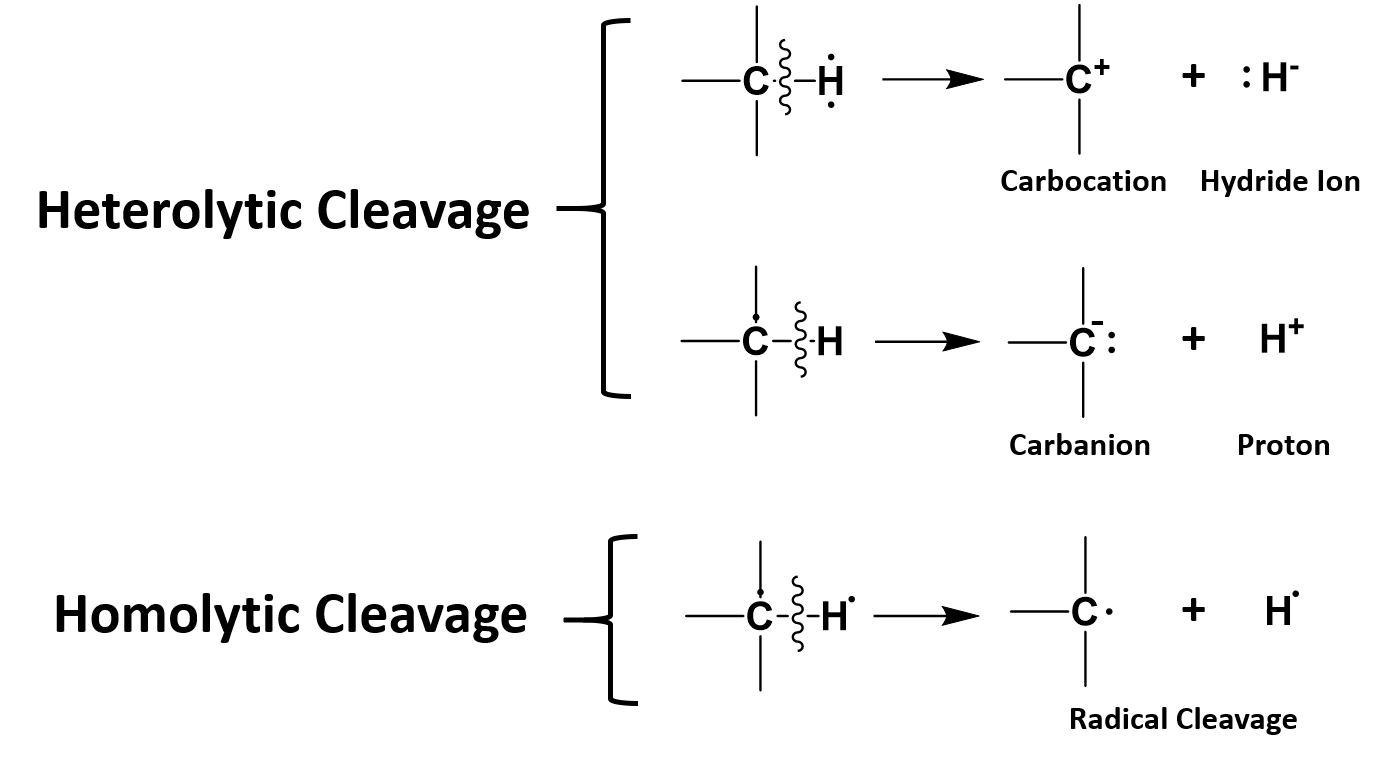
Figure 7.2 Examples of Heterolytic and Homolytic Bond Cleavage. In heterolytic cleavage both shared electrons are retained by one of the atoms from the shared bond, whereas in homolytic cleavage one electron is retained by each atom of a shared bond forming radical intermediates.
Nucleophiles and Electrophiles
A nucleophile is an atom or functional group with a pair of electrons (usually a non-bonding, or lone pair) that can be shared. The same, however, can be said about a base: in fact, bases can act as nucleophiles, and nucleophiles can act as bases. What, then, is the difference between a base and a nucleophile?
A Brønsted-Lowry base uses a lone pair of electrons to form a new bond with an acidic proton. Remember that when we evaluate basicity – the strength of a base – we speak in terms of thermodynamics: where does equilibrium lie in a reference acid-base reaction?
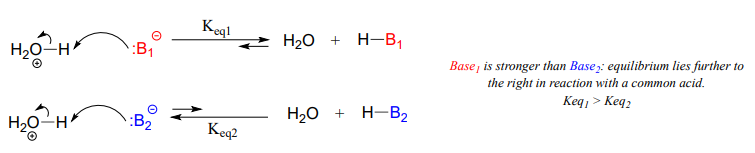
Figure 7.3 Brønsted-Lowry Bases. The upper diagram and lower diagram show the equilibrium positions of strong and weak bases, respectively.
A nucleophile shares its lone pair of electrons with an electrophile – an electron-poor atom other than a hydrogen, usually a carbon. When we evaluate nucleophilicity, we are thinking in terms of kinetics – how fast does the nucleophile react with a reference electrophile?
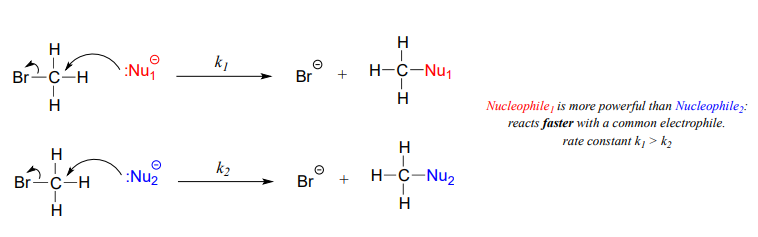
Figure 7.4 Nucleophiles. The upper and lower diagrams show faster reacting and slower reacting nucleophiles, respectively.
In both laboratory and biological organic chemistry, the most common nucleophilic atoms are oxygen, nitrogen, and sulfur, and the most common nucleophilic compounds and functional groups are water/hydroxide ion, alcohols, phenols, amines, thiols, and sometimes carboxylates.
Under certain circumstances carbon atoms can also act as nucleophiles. For example, enolate ions are common carbon nucleophiles in biochemical reactions, while the cyanide ion (CN−) is an example of a carbon nucleophile commonly used in the laboratory (Figure 7.5).
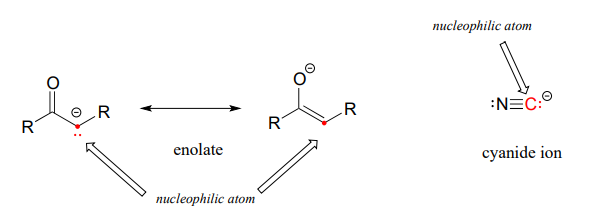
Figure 7.5 Carbon Can Act as a Nucleophile Under Certain Circumstances.
Many of the enzymatic functions performed in living systems use proteins as the catalyst. However, some enzymatic reactions can be mediated by other macromolecules such as RNA and will be discussed in more detail at the end of this chapter. Within protein structures, several of the amino acid R-groups can serve as nucleophiles and are often found in the active site of enzymes. These include: Cysteine, Serine, Threonine, Tyrosine, Glutamic Acid, Aspartic Acid, Lysine, Arginine, and Histidine (Figure 7.6).
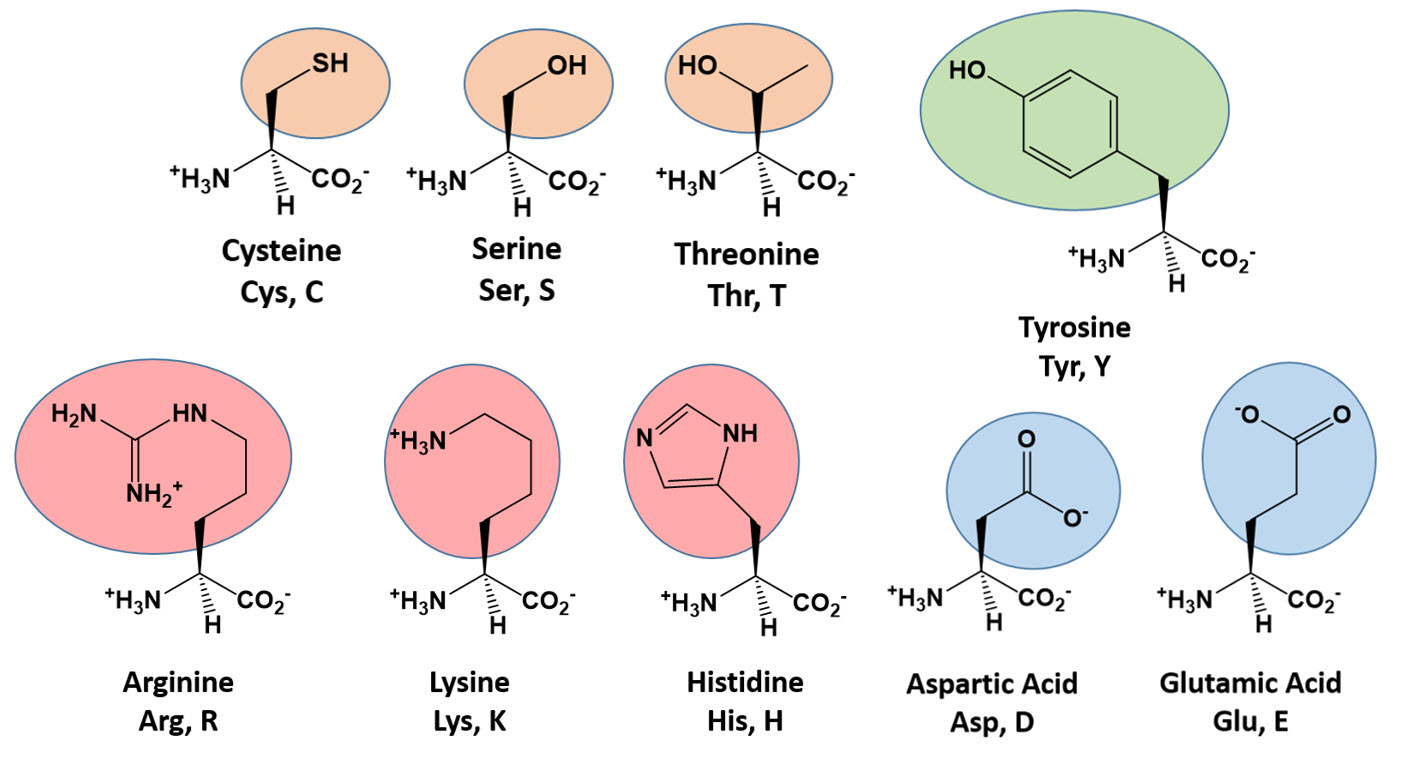
Figure 7.6 Amino Acid R-Groups that Commonly Act as a Nucleophile. Reactive amino acids, that can serve as a nucleophile, are often found in the active site of enzymes and participate in the reaction mechanism.
Electron Pushing
Arrow pushing or electron pushing is a technique used to describe the progression of organic chemistry reaction mechanisms. It was first developed by Sir Robert Robinson. In using arrow pushing, “curved arrows” or “curly arrows” are superimposed over the structural formulae of reactants in a chemical equation to show the reaction mechanism. The arrows illustrate the movement of electrons as bonds between atoms are broken and formed. Arrow pushing is also used to describe how positive and negative charges are distributed around organic molecules through resonance. It is important to remember, however, that arrow pushing is a formalism and electrons (or rather, electron density) do not move around so neatly and discretely in reality.
Organic chemists use two types of arrows within molecular structures to describe electron movements. Single electrons’ trajectories are designated with single barbed arrows, whereas double-barbed arrows show movement of electron pairs (Figure 7.7).
![]()
Figure 7.7 Trajectory of electron pairs (upper diagram) and the trajectory of single electron movement (lower diagram).
Figure from: Jkwchui
When a bond is broken, electrons leave where the bond was; this is represented by a curved arrow pointing away from the bond and an ending the arrow pointing towards the next unoccupied molecular orbital. Similarly, organic chemists represent the formation of a bond by a curved arrow pointing between two species.
For clarity, when pushing arrows, it is best to draw the arrows starting from a lone pair of electrons or a σ or π bond and ending in a position that can accept a pair of electrons, allowing the reader to know exactly which electrons are moving and where they are ending.
Acid Dissociation Constants, Ka and pKa
The Brønsted-Lowry definition of an acid is any species that can donate a proton, whereas a base is any species that can accept a proton. The strength of an acid is measured in terms of its potential to dissociate within a solution and is characterized by a specific acid dissociation constant, Ka, (also known as acidity constant, or acid-ionization constant). The Ka is a quantitative measure of the strength of an acid in solution and is the equilibrium constant for a chemical reaction

known as dissociation in the context of acid–base reactions. The chemical species HA is an acid that dissociates into A−, the conjugate base of the acid and a hydrogen ion, H+. The system is said to be in equilibrium when the concentrations of its components will not change with the passing of time, because both forward and backward reactions are occurring at the same rate.
The dissociation constant is defined by
![{\displaystyle K_{\mathrm {a} }=\mathrm {\frac {[A^{-}][H^{+}]}{[HA]}} }](https://wikimedia.org/api/rest_v1/media/math/render/svg/4df1424aa532585fc7ad6dd660a596705ee31a2b)
- or
![{\displaystyle pK_{{\ce {a}}}=-\log _{10}K_{a}=\log _{10}{\frac {{\ce {[HA]}}}{[{\ce {A^-}}][{\ce {H+}}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ac7c3fee703f954616c35979152974a9a3356537)
where quantities in square brackets represent the concentrations of the species at equilibrium.
The acid dissociation constant is a direct consequence of the underlying thermodynamics of the dissociation reaction. The pKa value is directly proportional to the standard Gibbs free energy change for the reaction. The value of the pKa changes with temperature and can be understood qualitatively based on Le Châtelier’s principle, as follows: when the reaction is endothermic, Ka increases and pKa decreases with increasing temperature; the opposite is true for exothermic reactions. The value of pKa also depends on the molecular structure of the acid.
The quantitative behavior of acids and bases in solution can be understood only if their pKa values are known. In particular, the pH of a solution can be predicted when the analytical concentration and pKa values of all acids and bases are known; conversely, it is possible to calculate the equilibrium concentration of the acids and bases in solution when the pH is known. These calculations find application in many different areas of chemistry, biology, medicine, and geology. For example, many compounds used for medication are weak acids or bases, and a knowledge of the pKa values, together with the water–octanol partition coefficient, can be used for estimating the extent to which the compound enters the blood stream. In living organisms, acid–base homeostasis and enzyme kinetics are dependent on the pKa values of the many acids and bases present in the cell and in the body.
Primary Enzyme Classifications
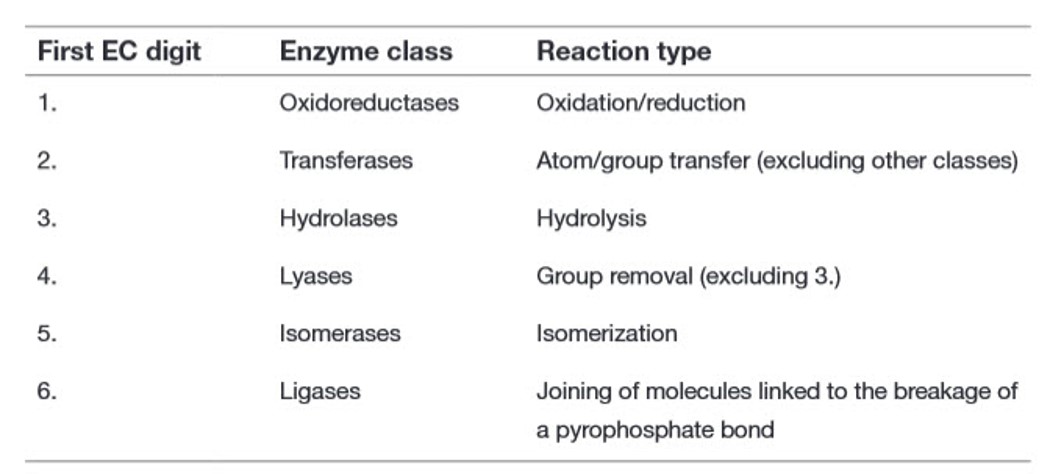
Recall from Chapter 6 that there are six major classes of biochemical reactions that are mediated by enzymes (Table 7.1). These include oxidation-reduction reactions, group transfer reactions, hydrolysis reactions, the formation/removal of carbon-carbon double bonds, isomerization reactions, and ligation reactions. This section will give you a brief introduction to these six types of reactions.
Table 7.1 Primary Enzyme Classification System by the Enzyme Commission (EC)
Oxidation-Reduction Reactions
An oxidation-reduction (redox) reaction is a type of chemical reaction that involves a transfer of electrons between two atoms or compounds. The substance that loses the electrons is said to be oxidized, while the substance that gains the electrons is said to be reduced. Redox reactions always have to occur together. If one molecule is oxidized, then another molecule has to be reduced (ie. electrons don’t appear out of thin air to be added to a compound, they always have to come from somewhere!).
The change in electron composition can be evaluated in the change of the oxidation state (or number) of an atom. Therefore, an oxidation-reduction reaction is any chemical reaction in which the oxidation state (number) of a molecule, atom, or ion changes by gaining or losing an electron. We will learn how to evaluate the oxidation state of a molecule within this section. Overall, redox reactions are common and vital to some of the basic functions of life, including photosynthesis, respiration, combustion, and corrosion or rusting.
As shown in Figure 7.8, an easy mnemonic for helping you remember which member gains electrons and which member loses electrons is ‘LEO the lion says GER’, where LEO stands for Lose Electrons = Oxidized and GER stands for Gain Electrons = Reduced.
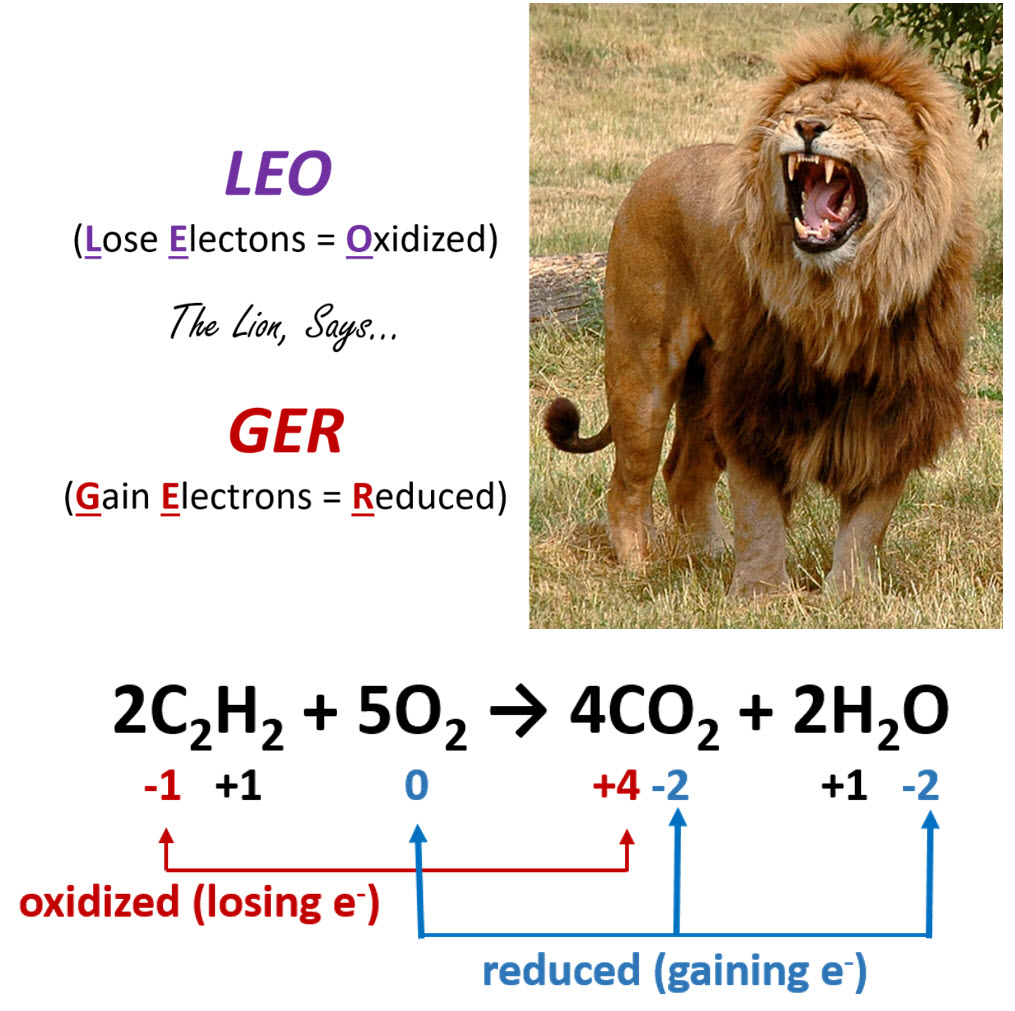
Figure 7.8. The Rules of Oxidation and Reduction. (Upper Diagram) The mnemonic LEO the lion says GER is a helpful way to remember the major concepts of Oxidation-Reduction reactions, noting that when a molecule Loses Elections it is Oxidized (LEO), and when a molecule Gains Electrons it is Reduced (GER). (Lower Diagram) An example of a redox reaction with the oxidized and reduced components indicated.
Lion photo modified from Robek
Group Transfer Reactions
In group transfer reactions, a functional group will be transferred from one molecule that serves as the donor molecule to another molecule that will be the acceptor molecule. The transfer of an amine functional group from one molecule to another is common example of this type of reaction and is shown in Figure 7.9 below.
![]()
Figure 7.9 Transfer of an Amine Functional Group. A common group transfer reaction in biological systems is one that is used to produce α-amino acids that can then be used for protein synthesis. In this reaction, one α-amino acid serves as the donor molecule and an α-keto acid (these molecules contain a carboxylic acid functional group and a ketone functional group separated by one α-carbon) serves as the acceptor. In the acceptor molecule, the carbonyl oxygen is replaced with the amine functional group, whereas in the donor molecule, the amine functional group is replaced by an oxygen forming a new ketone functional group.
Hydrolysis Reactions
The classification of hydrolysis reactions include both the forward reactions that involve the addition of water to a molecule to break it apart or the reverse reaction involving the removal of water to join molecules together, termed dehydration synthesis (or condensation) (Figure 7.7). When water is added to a molecule to break it apart into two molecules this reaction is called hydrolysis. The term ‘lysis‘ means to break apart, and the term ‘hydro‘ refers to water. Thus, the term hydrolysis means to break apart with water. The reverse of that reaction involves the removal of water from two molecules to join them together into a larger molecule. Since the two molecules are losing water, they are being dehydrated. Thus, the formation of molecules through the removal of water is known as dehydration synthesis. Since water is also a by-product of these reactions, they are also commonly referred to as condensation reactions. As we have seen in Chapter 6, the formation of the major classes of macromolecules in the body (proteins, carbohydrates, lipids, and nucleic acids) are formed through dehydration synthesis where water is removed from the molecules (Figure 7.10). During normal digestion of our food molecules, the major macromolecules are broken down into their building blocks through the process of hydrolysis.
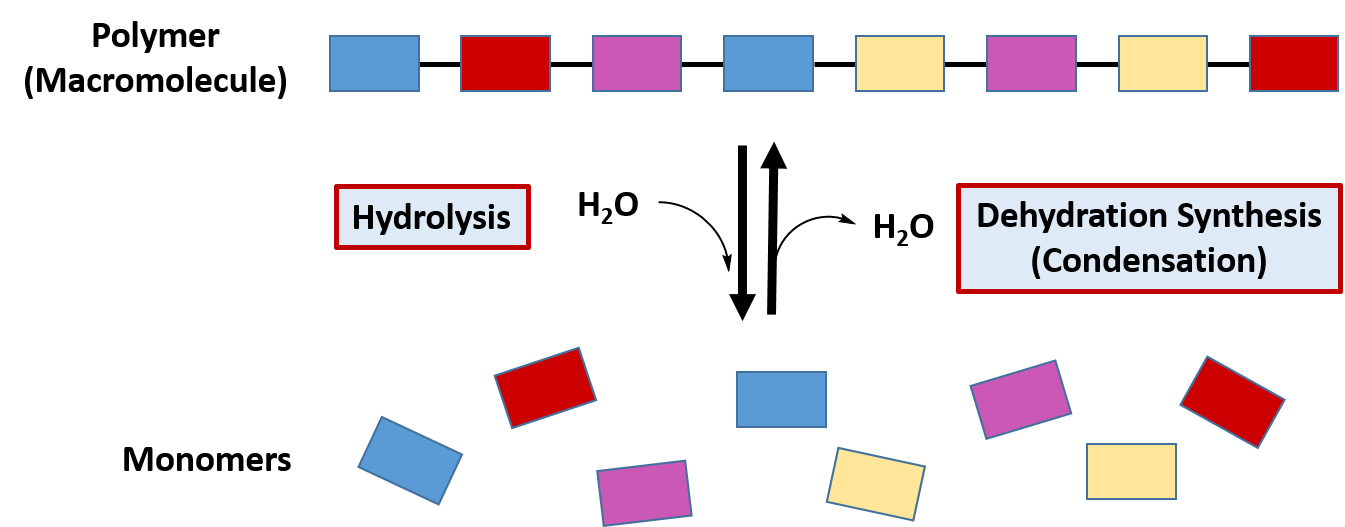
Figure 7.10 Hydrolysis and Dehydration Synthesis. The reactions of hydrolysis mediate the breakdown of larger polymers into their monomeric building blocks by the addition of water to the molecules. The reverse of the reaction is dehydration synthesis, where water is removed from the monomer building blocks to create the larger polymer structure.
As you learned throughout this term, the major macromolecules are built by putting together repeating monomer subunits through the process of dehydration synthesis. Interestingly, the organic functional units used in the dehydration synthesis processes for each of the major types of macromolecules have similarities with one another. Thus, it is useful to look at the reactions together (Figure 7.11)
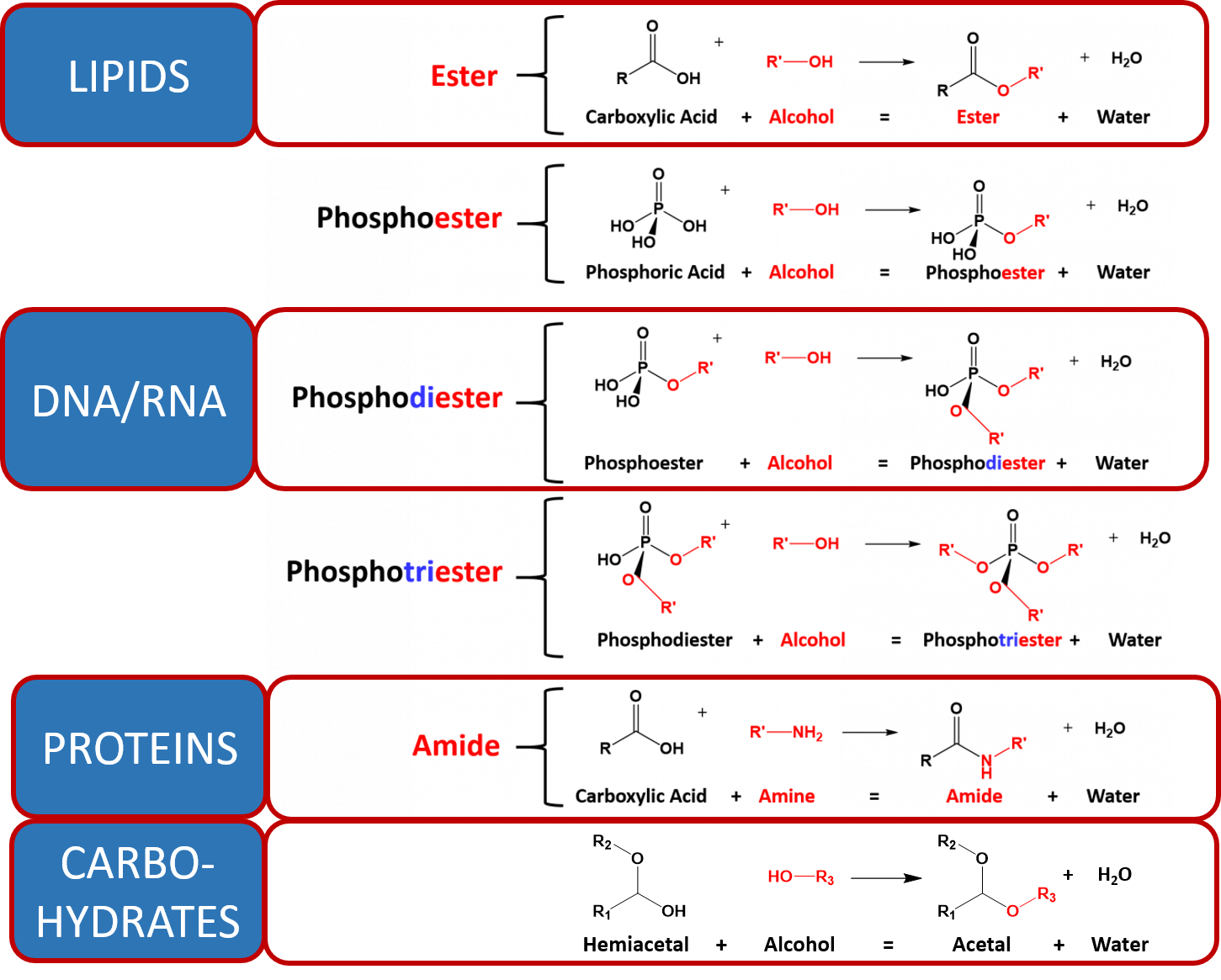
Figure 7.11 Dehydration Synthesis Reactions Involved in Macromolecule Formation. The major organic reactions required for the biosynthesis of lipids, nucleic acids (DNA/RNA), proteins, and carbohydrates are shown. Note that in all of the reactions, there is a functional group that contains two electron withdrawing groups (the carboxylic acid, phosphoric acid and the hemiacetal each have two oxygen atoms attached to a central carbon or phosphorus atom). This forms a reactive partially positive center atom (carbon in the case of the carboxylic acid and hemiacetal, or phosphorus in the case of the phosphoric acid) that can be attacked by the electronegative oxygen or nitrogen from an alcohol or amine functional group.
The Formation/Removal of Carbon-Carbon Double Bonds
Reactions that mediate the formation and removal of carbon-carbon double bonds are also common in biological systems and are catalyzed by a class of enzymes called lyases. The formation or removal of carbon-carbon double bonds is also used in synthetic organic chemistry reactions to create desired organic molecules. One of these types of reactions is called a hydrogenation reaction, where a molecule of hydrogen (H2) is added across a C-C double bond, reducing it to a C-C single bond. If this is done using unsaturated oils, the unsaturated fats can be converted into saturated fats (Figure 7.12). This type of reaction is commonly done to produce partially hydrogenated oils converting them from liquids at room temperature into solids. Margarines made from vegetable oil are made in this manner. Unfortunately, a by-product of this reaction can be the formation of TAGS containing trans double bonds. Once the health hazards of consuming trans fats was recognized, the Food and Drug Administration (FDA) placed a ban on the inclusion of trans fats in food products. This ban was enacted in the summer of 2015 and gave food-makers three years to eliminate them from the food supply, with a deadline of June 18, 2018.

Figure 7.12 Hydrogenation of Oils to Produce Margarine. Unsaturated oils can by partially or fully hydrogenated to produce the saturated fatty acids to produce margarines that will remain solid at room temperature. The addition of the new hydrogen atoms to create the saturated hydrocarbons are shown in yellow in the final product.
Upper photo provided by Cottonseed Oil and lower photo provided by Littlegun
Isomerization Reactions
In isomerization reactions a single molecule is rearranged such that it retains the same molecular formula but now has a different bonding order of the atoms forming a structural or stereoisomer. The conversion of glucose 6-phosphate to fructose 6-phosphate is a good example of an isomerization reaction and is shown in figure 7.13
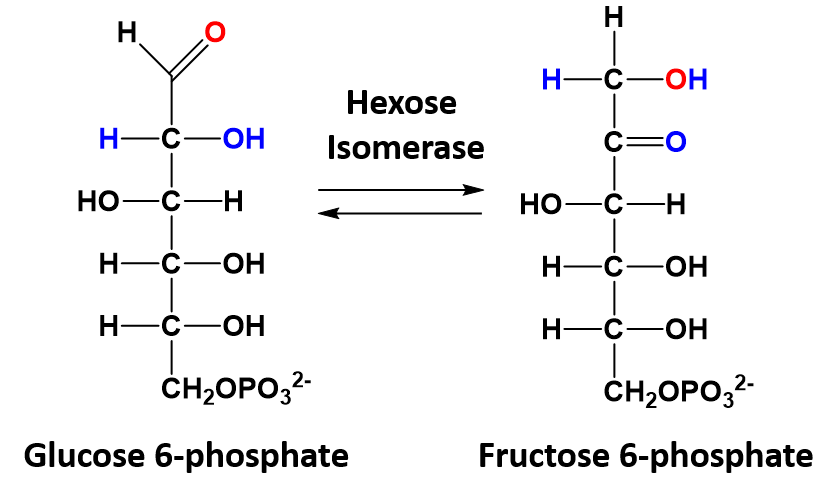
Figure 7.13 Isomerization of Glucose 6-phosphate to Fructose 6-phosphate.
Ligation Reactions
Ligation reactions use the energy of ATP to join two molecules together. An example of this kind of reaction is the joining of the amino acid with the transfer RNA (tRNA) molecule during protein synthesis. During protein synthesis the tRNA molecules bring each of the amino acids to the ribosome where they can be incorporated into the newly growing protein sequence. To do this, the tRNA molecules must first be attached to the appropriate amino acid. Specific enzymes are available called amino acyl – tRNA synthetases that mediate this reaction. The synthetase enzymes use the energy of ATP to covalently attach the amino acid to the tRNA molecule. A diagram of this process is shown in Figure 7.14. For each of the 20 amino acids, there is a specific tRNA molecule and a specific synthetase enzyme that will ensure the correct attachment of the correct amino acid with its tRNA molecule.
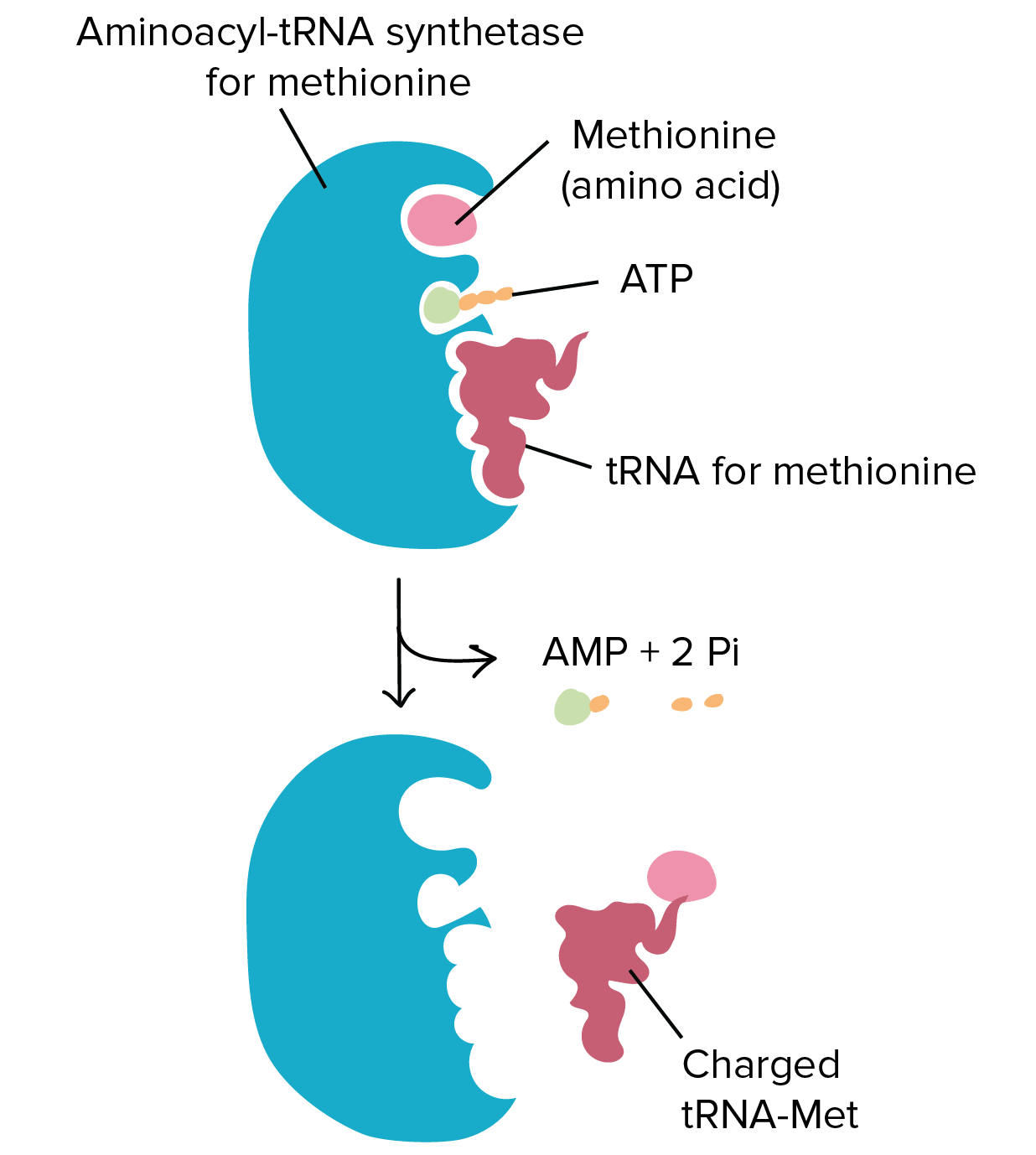
Figure 7.14 Ligation Reaction Covalently Attaching Methionine with the Appropriate tRNA. The amino-acyl tRNA synthetase enzyme for methionine (shown in blue) covalently attaches methionine (light pink) with the methionine tRNA molecule (dark pink). This reaction requires the energy provided from the breakdown of the ATP molecule into AMP, releasing energy with the breakdown of the phosphate bonds into two inorganic phosphate ions (2 Pi).
Figure provided by the Kahn Academy
7.2 Overview of Catalytic Mechanisms
The descriptions below describe the major mechanisms enzymes use to catalyze chemical reactions. It should be noted that many enzymes use more than one, and sometimes several different catalytic strategies during their reaction mechanism.
Covalent Catalysis
Covalent catalysis involves the formation of a covalent bond between the enzyme and at least one of the substrates involved in the reaction. Often times this involves nucleophilic catalysis which is a subclass of covalent catalysis. As seen in Section 7.1, several amino acid R-groups can serve as a nucleophile and are often found at the active site of enzymes. Nucleophilic side chains are often activated by deprotonation caused by neighboring side chains, such as histidine that can act as a base. Alternatively, water can also activate the nucleophile. The intermediate covalent bond formation between the enzyme and the substrate enables bond cleavage and the removal of a leaving group.
Acid-Base Catalysis
Acid-Base Catalysis is involved in any reaction mechanism that requires the transfer of a proton from one molecule to another. It is very common to see this mechanism combined with Covalent Catalysis as many nucleophiles are activated by the removal of a proton, including alcohol, thiol, and amine functional groups. Enzymes that utilize Acid-Base Catalysis can be subgrouped further into either specific acid-base or general acid-base reactions. Specific acid or specific base catalysis occur if a hydronium ion (H3O+) or a hydroxide ion (OH–), respectively, are utilized directly in the reaction mechanism, and the pH of the solution affects the rate of catalysis. General acid and general base reactions occur when molecules other than hydronium ion (H3O+) or a hydroxide ion (OH–) are the source of proton donation or acceptance. Most commonly, an active site amino acid residue is used to accept or donate a proton within the reaction mechanism. In general acid-base reactions the pH is usually held constant within a buffered system.
Electrostatic Catalysis
Electrostatic catalysis occurs when the enzyme active site stabilizes the transition state of the reaction by forming electrostatic interactions with the substrate. The electrostatic interactions can be ionic, ionic-dipole, dipole-dipole, or hydrophobic interactions. Hydrogen bonding is one of the most common electrostatic interactions formed in the active site.
Desolvation
Enzyme active sites can become devoid of water and mimic the reaction characteristics of the gas phase. This can destabilize the polarized state of charged groups such as acids and bases. Thus, the neutral form of these types of residues becomes the favored state. This is due to significant alterations in the pKa of the active site residues within the nonpolar environment. This can cause normally acidic residues such as glutamate to abstract a proton from histidine and behave as a base, for example.
Catalysis by Approximation
In catalysis by approximation, the enzyme enhances the reaction rate by binding with multiple substrates and positioning them favorably so that the reaction can proceed. Binding with the enzyme reduces the rotational entropy of the substrates that would otherwise be randomly free floating in solution, and enables the correct positioning of substrates for the reaction. The loss of entropy, which is not favorable, is offset by the binding energy released with the substrate-enzyme interaction.
In addition to correctly positioning the substrates to interact with one another, catalysis by approximation converts a reaction that would have been second order, with substrates that are free floating in solution, to a first order reaction, where all of the substrates are held in place by the enzyme and behave as a single molecule. This can dramatically improve the catalytic rate of the reaction from 105 to 107 times faster, depending on the enzyme system.
Strain Distortion
In organic chemistry, you learned that certain structures such as three-membered and four-membered ring structures, such as epoxides were highly reactive due to the strain distortion inherent to the unfavored bond angles inherent to the ring. Enzyme active sites can also utilize strain distortion within a bound substrate to increase the reactivity of the molecule and favor the formation of the transition state. Many enzymes that function by the induced fit model also utilize strain distortion within their catalytic mechanism. Within the unbound state they remain in a low catalytic state, however the interaction with the substrate induces the destabilization of the enzyme active site or may induce strain within the substrate causing the initiation of the catalytic activity of the enzyme.
Cofactor Catalysis
Cofactors are molecules that bind to enzymes and are required for the catalytic activity of the enzyme. They can be divided into two major categories: metals and coenzymes. Metal cofactors that are commonly found in human enzymes include: iron, magnesium, manganese, cobalt, copper, zinc, and molybdenum. Coenzymes are small organic molecules that are often derived from vitamins, which are essential organic nutrients consumed within the diet. Coenzymes can bind loosely with the enzyme and have the ability to bind and release from the active site, or they may be tight binding and lack the ability to release easily from the enzyme. Tight binding coenzymes are referred to as prosthetic groups. Enzymes that are not yet associated with a required cofactor are called apoenzymes, whereas enzymes that are bound with their required cofactors are called holoenzymes. Sometimes organic molecules and metals combine to form coenzymes, such as in the case of the heme cofactor (Figure 7.15). Coordination of heme cofactors with their enzyme counterparts often involves electrostatic interactions with histidine residues as shown in the succinate dehydrogenase enzyme shown in Figure 7.15.
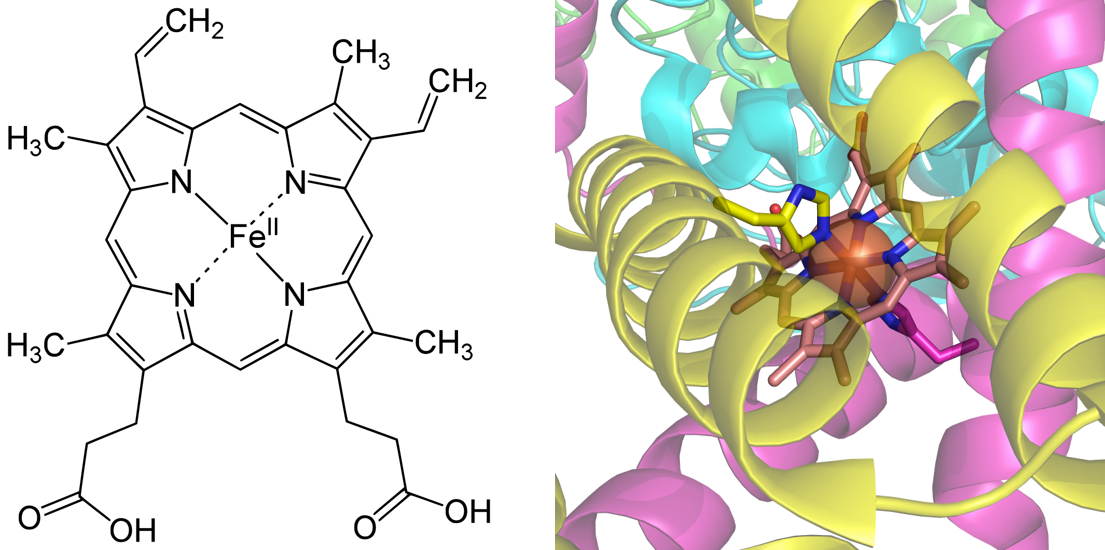
Figure 7.15 The Heme Cofactor. The family of heme cofactors contain an iron metal coordinated with a porphyrin ring structure as shown in the left hand panel within the structure of Heme B. In the right hand panel, Heme B is shown complexed with the succinate dehydrogenase enzyme from the Kreb Cycle.
Structure of Heme B shown in the left hand panel is from: Yikrazuul and the crystal structure of Succinate Dehydrogenase complexed with Heme B is from: Richard Wheeler.
As an example of how vitamins can be utilized as cofactors, Table 7.2 shows the common B vitamins and the coenzymes derived from their structures. Many vitamin deficiencies cause disease states due to the inactivity of apoenzymes that are unable to function without the correctly bound coenzyme.
Table 7.2 Essential B-Vitamins and their Modified Enzyme Cofactors
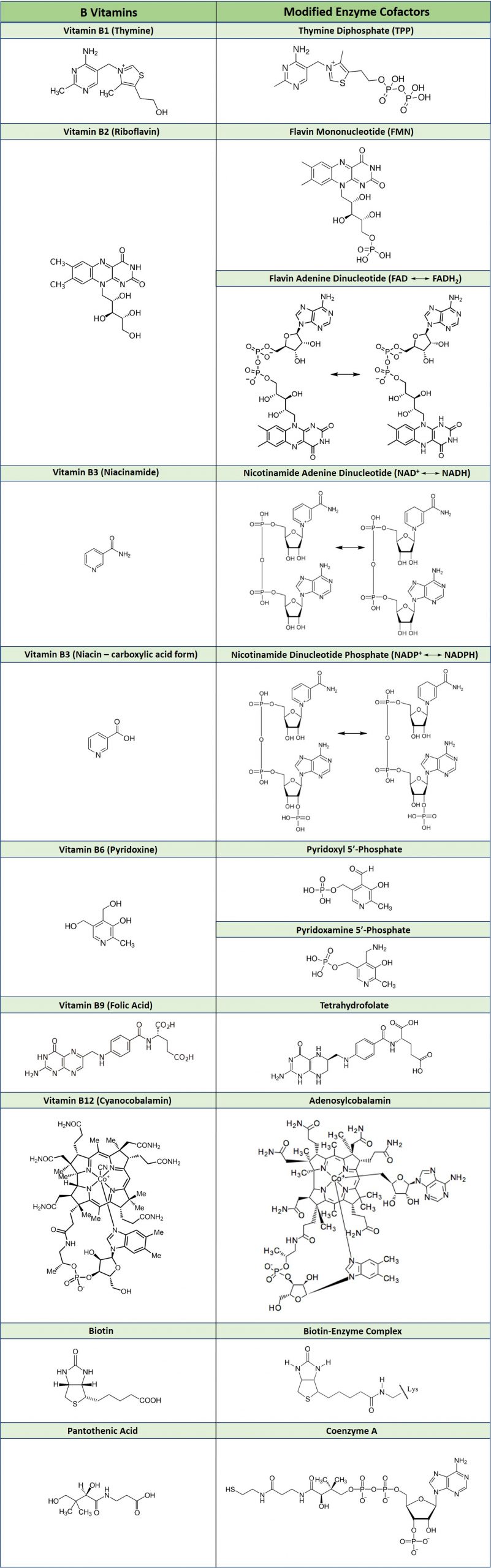
Cofactors can help to mediate enzymatic reactions through the use of any of the different catalytic strategies listed above. They can serve as nucleophiles and mediate covalent catalysis or form electrostatic interactions with the substrate and stabilize the transition state. They can also cause strain distortion or facilitate acid-base catalysis. Metal-aided catalysis can often use homolytic reaction mechanisms that involve radical intermediates. This can be important in reactions such as those occurring in the electron transport chain that require the safe movement of single electrons.
7.3 Examples of Reaction Mechanisms
Protease Enzymes
Proteolytic enzymes (also termed peptidases, proteases and proteinases) are capable of hydrolyzing peptide bonds in proteins. They can be found in all living organisms, from viruses to animals and humans. Proteolytic enzymes have great medical and pharmaceutical importance due to their key role in biological processes and in the life-cycle of many pathogens. Proteases are extensively applied enzymes in several sectors of industry and biotechnology, furthermore, numerous research applications require their use, including production of Klenow fragments, peptide synthesis, digestion of unwanted proteins during nucleic acid purification, cell culturing and tissue dissociation, preparation of recombinant antibody fragments for research, diagnostics and therapy, and the exploration of the structure-function relationships.
Proteolytic enzymes belong to the hydrolase class of enzymes and are grouped into the subclass of the peptide hydrolases or peptidases. Depending on the site of enzyme action the proteases can also be subdivided into exopeptidases or endopeptidases. Exopeptidases, such as aminopeptidases and carboxypeptidases catalyze the hydrolysis of the peptide bonds near the N– or C-terminal ends of the substrate, respectively (Figure 7.16). Endopeptidases (Figure 7.16) cleave peptide bonds at internal locations within the peptide sequence. Proteases may also be nonspecific and cleave all peptide bonds equally or they may be highly sequence specific and only cleave peptides after certain residues or within specific localized sequences.
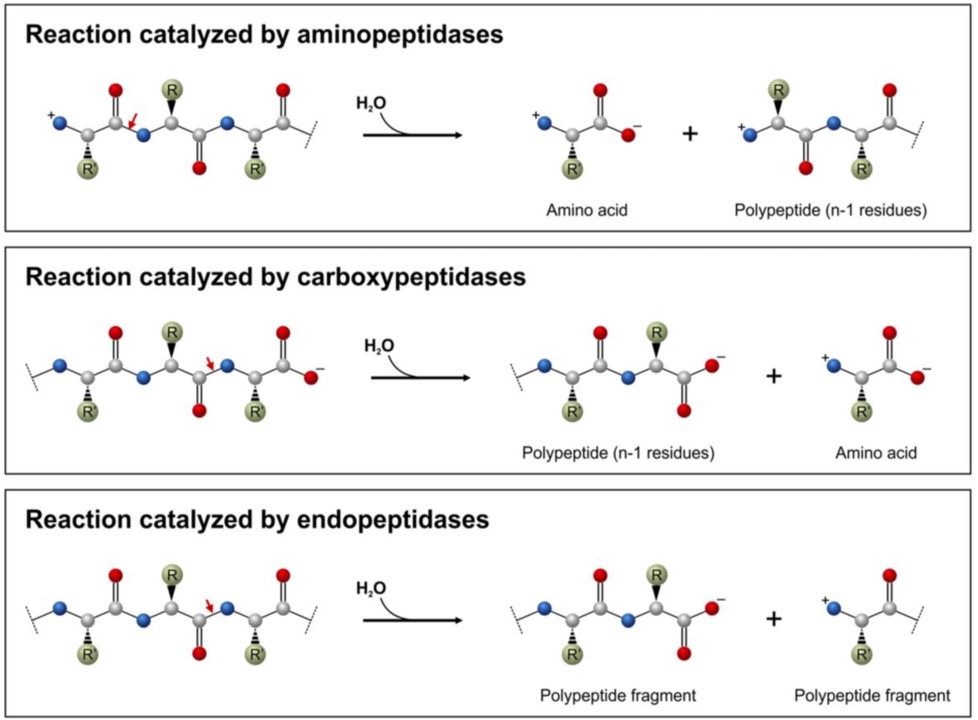
Figure 7.16 Common Peptidase Reactions. Aminopeptidases (top diagram) and carboxypeptidases (middle diagram) remove the terminal amino acid residues, whereas endopeptidases (lower diagram) cleave protein sequences at internal sites. The red arrows show the peptide bonds to be cleaved.
Figure from: Mótyán, J.A., et al. (2013) Biomolecules 3(4), 923-942
The action of proteolytic enzymes is essential in many physiological processes. For example, proteases function in the digestion of food proteins, protein turnover, cell division, the blood-clotting cascade, signal transduction, processing of polypeptide hormones, apoptosis and the life-cycle of several disease-causing organisms including the replication of retroviruses such as the human immunodeficiency virus (HIV). Due to their key role in the life-cycle of many hosts and pathogens they have great medical, pharmaceutical, and academic importance.
It was estimated previously that about 2% of the human genes encode proteolytic enzymes and due to their necessity in many biological processes, proteases have become important therapeutic targets. They are intensively studied to explore their structure-function relationships, to investigate their interactions with the substrates and inhibitors, to develop therapeutic agents for antiviral therapies or to improve their thermostability, efficiency and to change their specificity by protein engineering for industrial or therapeutic purposes.
Based on the catalytic mechanism and the presence of amino acid residue(s) at the active site the proteases can be grouped as aspartic proteases, cysteine proteases, glutamic proteases, metalloproteases, asparagine proteases, serine proteases, threonine proteases, and proteases with mixed or unknown catalytic mechanism. Here we will explore the reaction mechanism and sequence specificity of the family of serine proteases.
The list of serine proteases is quite long. They are grouped in two broad categories – 1) those that are chymotrypsin-like and 2) those that are subtilisin-like. Though subtilisin-type and chymotrypsin-like enzymes use the same mechanism of action, including the catalytic triad, the enzymes are otherwise not related to each other by sequence and appear to have evolved independently (Figure 7.17). They are, thus, an example of convergent evolution – a process where evolution of different forms converge on a structure to provide a common function.
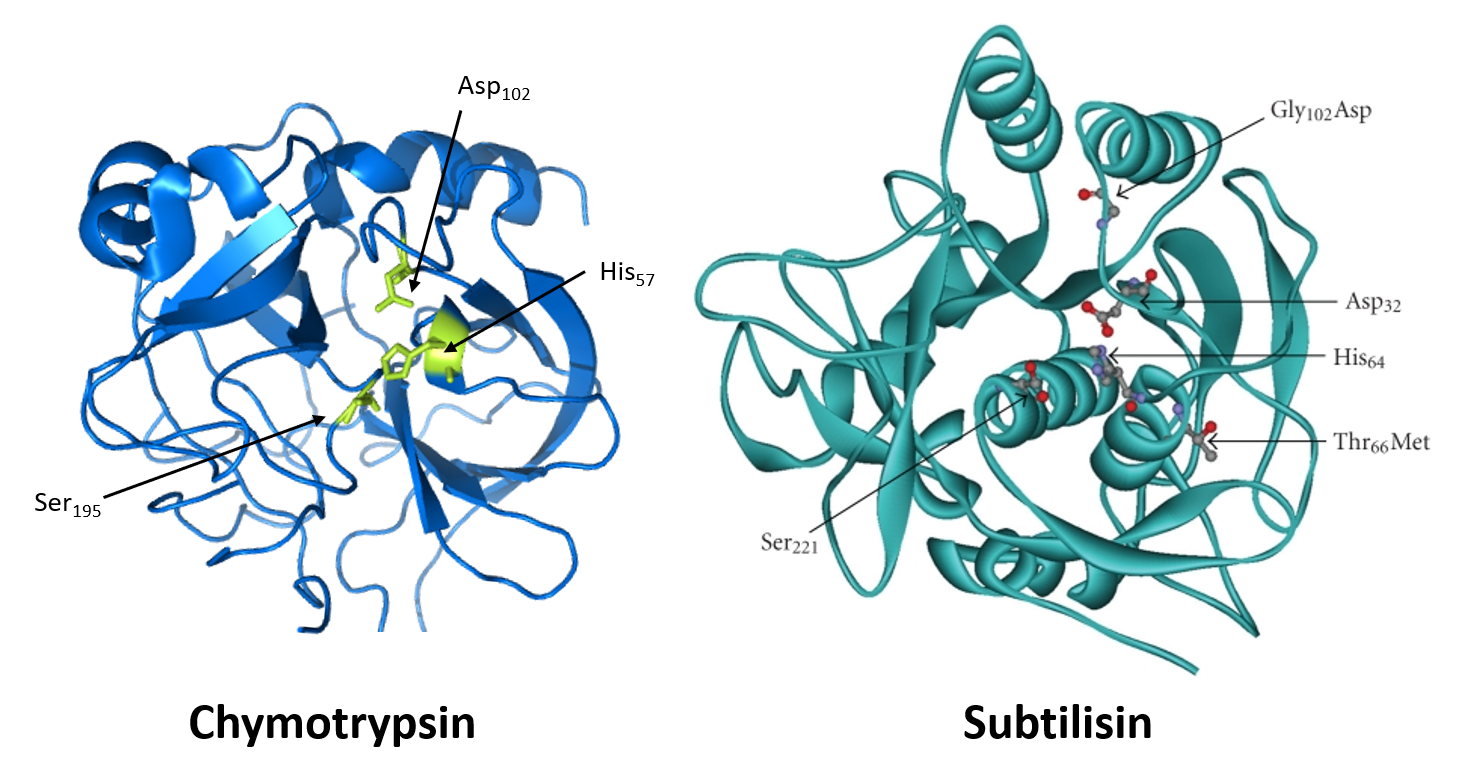
Figure 7.17 Convergent Evolution of the Serine Proteases, Chymotrypsin and Subtilisin. The crystal structure of the eukaryotic, Bovine Chymotrypsin (left hand panel) with catalytic triad indicated in green. The crystal structure of the prokaryotic Subtilisin BPN from the bacterium Bacillus subtilis (right hand panel) with the catalytic triad and common mutations indicated using the ball and stick models. Note that the catalytic triad formation is strikingly similar between the two structures, whereas the surrounding protein structures and sequence do not show homology or related ancestry.
Image of bovine chymotrypsin modified from Mattyjenjen and image of subtilisin BNP from Romero-Garcia, E.R., et al. (2009) J Biomed Biotech 2009(1):201075
The chymotrypsin-like serine protease enzymes cleave the peptide bond on the carboxylic acid side of specific amino acids and the specificity is determined by the size/shape/charge of amino acid side chain that fits into the enzyme’s S1 binding pocket (Figure 7.18). Three chymotrypsin-like family members that share high sequence homology are the pancreatic digestive enzymes, trypsin, chymotrypsin and elastase. The protein cleavage sites of these enzymes varies. Trypsin cleaves proteins on the carboxylic side of basic residues, such as lysine and arginine, while Chymotrypsin cleaves after aromatic hydrophobic amino acids, such as phenylalanine, tyrosine, and tryptophan, and Elastase cleaves after small, hydrophobic residues, such as glycine, alanine, and valine. As shown in Figure 7.18, variations in the amino acid residues within the binding pocket of these proteases, enables electrostatic interactions with the substrate and determines sequence specificity.
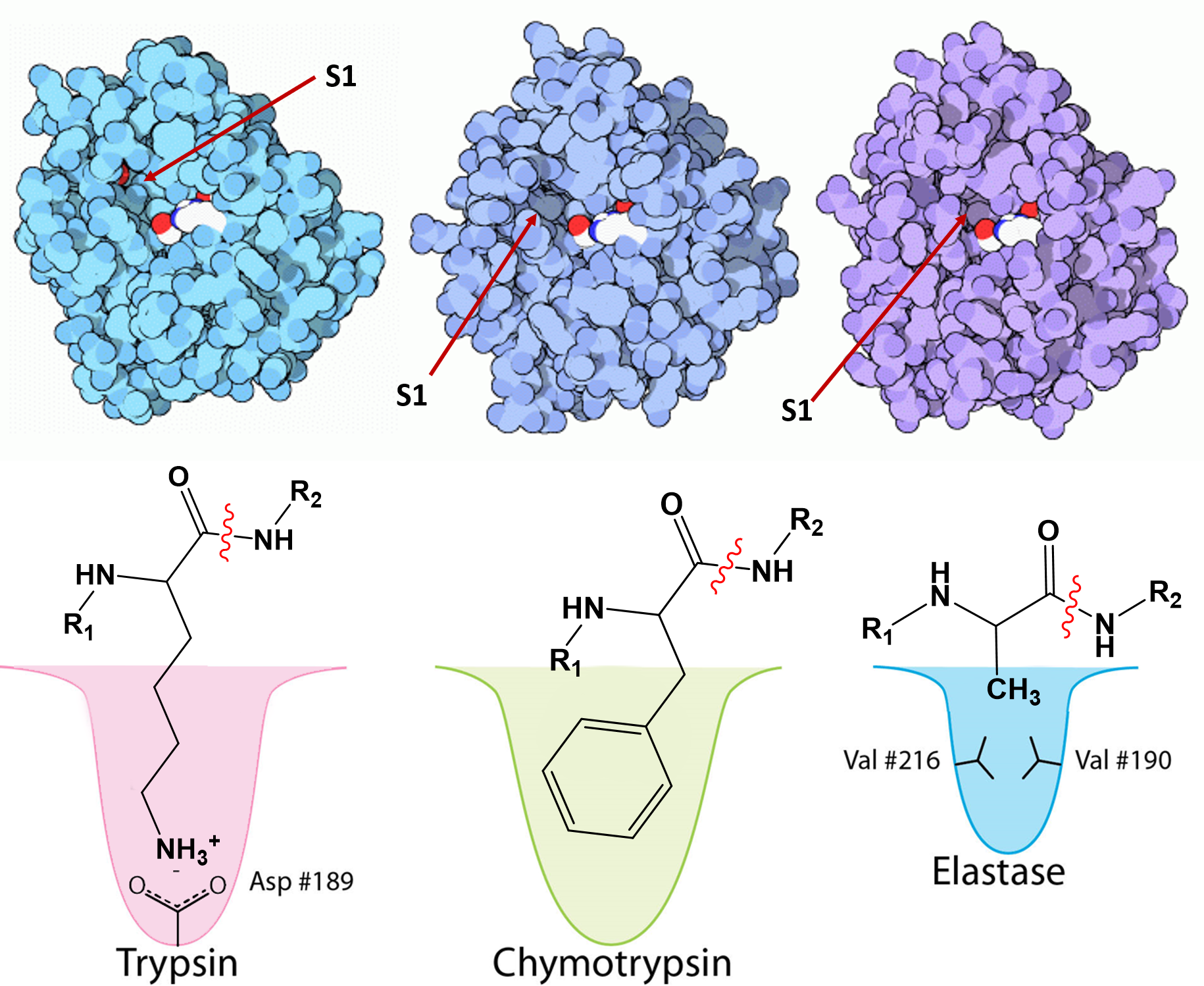
Figure 7.18 Substrate Specificity of Trypsin, Chymotrypsin, and Elastase. The upper panel shows the space-filling crystal structures of Trypsin, Chymotrypsin, and Elastase, respectively, with the S1 substrate binding pocket indicated. The lower panel depicts the S1 binding domains of each protease in more detail with important amino acid R-groups indicated. For Trypsin, an aspartate residue in the lower portion of the S1 pocket aid in electrostatic interactions with basic residues of the substrate. The Chymotrypsin S1 binding pocket is large and hydrophobic in nature accommodating aromatic residues of the substrate, while the Elastase S1 binding pocket is small and hydrophobic, only allowing other small and hydrophobic R-groups to dock in this location.
Image modified from: Goodsell, D. (2012) Molecule of the Month, Protein Database and Aleia Kim
Serine proteases use four of the major catalytic mechanism during the reaction cycle: Acid-Base Catalysis, Covalent Catalysis, Electrostatic Interactions, and Desolvation. The active site of serine proteases contains a catalytic triad of three amino acids: His, Ser (hence the name “serine protease”) and Asp. These three key amino acids each play an essential role in the cleaving ability of the proteases. While the amino acid members of the triad are located far from one another in the primary sequence of the protein, due to folding, they will be very close to one another in the heart of the enzyme.
During a catalysis event, an ordered mechanism occurs in which several intermediates are generated. The catalysis of the peptide cleavage can be seen as a ping-pong catalysis, in which a substrate binds (in this case, the polypeptide being cleaved), a product is released (the N-terminus “half” of the peptide), another substrate binds (in this case, water), and another product is released (the C-terminus “half” of the peptide). Figure 7.19 details the catalytic process.
In step 1 to 2, the polypeptide substrate enters the active site and is positioned in the correct orientation near the active site serine residue through electrostatic interactions in the S1 binding pocket. The carbonyl carbon of the substrate is positioned near the active site serine residue. The hydrogen of the serine alcohol is abstracted by the catalytic histidine residue through acid-base catalysis. This is made possible by the action of the active site aspartate residue. In this case, the aspartate residue abstracts a proton from histidine, enabling histidine to remove the proton from the serine alcohol. Normally, this would be an odd thing for aspartate to be able to do, as the pKa of the R-group of aspartate is much lower than that of histidine within an aqueous environment. However, when the peptide substrate docks in the active site of the enzyme, this excludes water from the area through a desolvation process, and creates a hydrophobic microenvironment. This effectively raises the pKa of aspartate and favors the uncharged or protonated form of the residue, causing the proton abstraction from histidine.
In steps 2 to 3 covalent catalysis is enabled as the active site serine mediates nucleophilic attack on the carbonyl carbon of the substrate forming a tetrahedral oxyanion intermediate. The oxyanion intermediate in the pathway is then stabilized by electrostatic interactions with a region of the protease known as the oxyanion hole. Here the negative charge of the oxyanion is stabilized by electrostatic interactions with amide nitrogens from the protease backbone. Rebounding of the oxyanion to reform the carbonyl double bond leads to the cleavage of the peptide bond. The C-terminal portion of the protein can then leave the active site of the enzyme.
Once the C-terminal peptide leaves the active site, water can enter and rehydrate the active site, as seen in steps 5 to 6. A water molecule is oriented in proximity to the carbonyl carbon of the N-terminal peptide that is still bound to the active site serine residue. The oxygen from water acts as a nucleophile and attacks the carbonyl carbon, recreating an oxyanion intermediate which is stabilized by the electrostatic interactions of the oxyanion hole. As this oxyanion rebounds to reform the carbonyl, the serine residue acts as a leaving group and the N-terminal peptide is released from the enzyme. The presence of water in the active site, re-establishes the serine residue and the native states of the histidine and aspartic acid residues.
Overall, each amino acid in the triad performs a specific task in this process:
- The serine has an -OH group that is able to act as a nucleophile, attacking the carbonyl carbon of the scissile peptide bond of the substrate (covalent catalysis).
- A pair of electrons on the histidine nitrogen has the ability to accept the hydrogen from the serine -OH group, thus coordinating the attack of the peptide bond (acid/base catalysis).
- The carboxyl group on the aspartic acid in turn coordinates with the histidine, making the nitrogen atom mentioned above much more electronegative through the process of desolvation.
Electrostatic interactions are critical to (1) bind the substrate in the S1 binding pocket (2) stabilize the transition state oxyanion, and (3) coordinate the water molecule to mediate nucleophilic attack on the enzyme-bound intermediate.
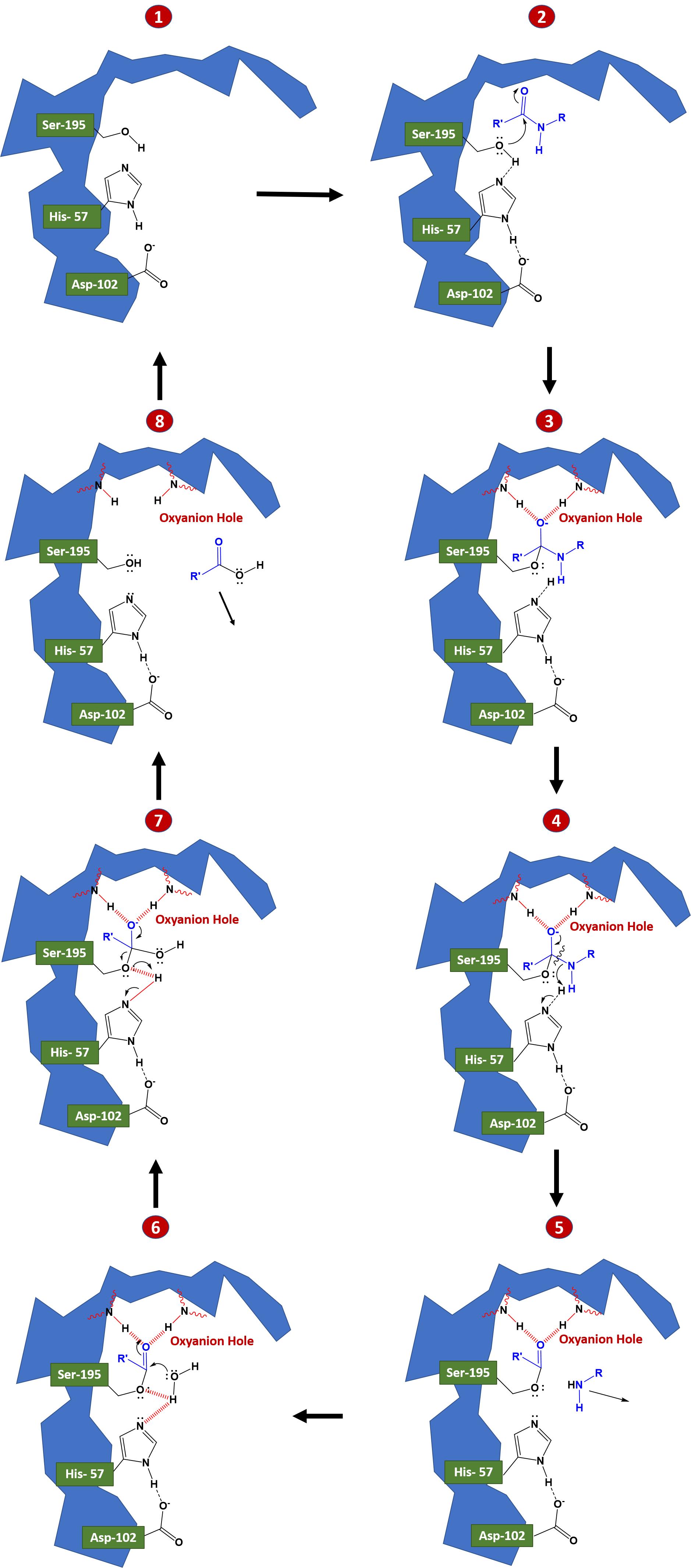
Figure 7.19 Reaction mechanism of Chymotrypsin-like Proteases. The reaction mechanism has been broken down into an eight step process. In steps 1-3 the protein substrate binds with the protease and is oriented to place the carbonyl carbon of the substrate in proximity with the active site serine residue. Acid-base catalysis enables the activation of the serine residue to mediate nucleophilic attack on the protein substrate. The covalent oxyanion intermediate shown in 3 and 4 is stabilized by the oxyanion hole. Rebound of the electrons to reform the carbonyl group cause the cleavage of the peptide bond and the removal of the C-terminal portion of the peptide from the active site, shown in 5. Water enters the active site and mediates nucleophilic attack on the carbonyl carbon of the enzyme-substrate intermediate, as shown in 6 and 7. As the carbonyl bond is reformed, serine acts as a leaving group and the N-terminal peptide is released from the enzyme. The catalytic triad in the enzyme active site is recovered and the enzyme is reset for another round of catalytic activity, as shown in 8.
Adenylate Kinases
Adenylate kinase (also known as AK or myokinase) is a phosphotransferase enzyme that catalyzes the interconversion of adenine nucleotides (ATP, ADP, and AMP). By constantly monitoring phosphate nucleotide levels inside the cell, AK enzymes play an important role in cellular energy homeostasis. The basic chemical reaction mediated by this enzyme class is the conversion of 2 ADP molecules into 1 ATP and 1 AMP (Figure 7.20A). The reverse reaction can also occur forming an equilibrium based on cellular concentrations of the varying phosphorylation states.
To date there have been nine human AK protein isoforms identified. While some of these are ubiquitous throughout the body, some are localized into specific tissues. For example, AK7 and AK8 are both only found in the cytosol of cells; and AK7 is found in skeletal muscle whereas AK8 is not. Not only do the locations of the various isoforms within the cell vary, but the binding of substrate to the enzyme and kinetics of the phosphoryl transfer are different as well. AK1, the most abundant cytosolic AK isozyme, has a Km about a thousand times higher than the Km of AK7 and 8, indicating a much weaker binding of AK1 to AMP. Sub-cellular localization of the AK enzymes is done by unique targeting sequences found in the protein. Each isoform also has different preference for NTP’s. Some will only use ATP, whereas others will accept GTP, UTP, and CTP as the phosphoryl carrier.
AK enzymes can be involved in regulating nucleotide concentrations and serve as a relay system between cellular and mitochondrial pools of adenine nucleotides, as shown in Figure 7.20B. AK enzymes can also serve as a sensor for energy load within the cell and can lead to the activation of AMP-sensitive systems within the cell when energy levels are low (Figure 7.20C).
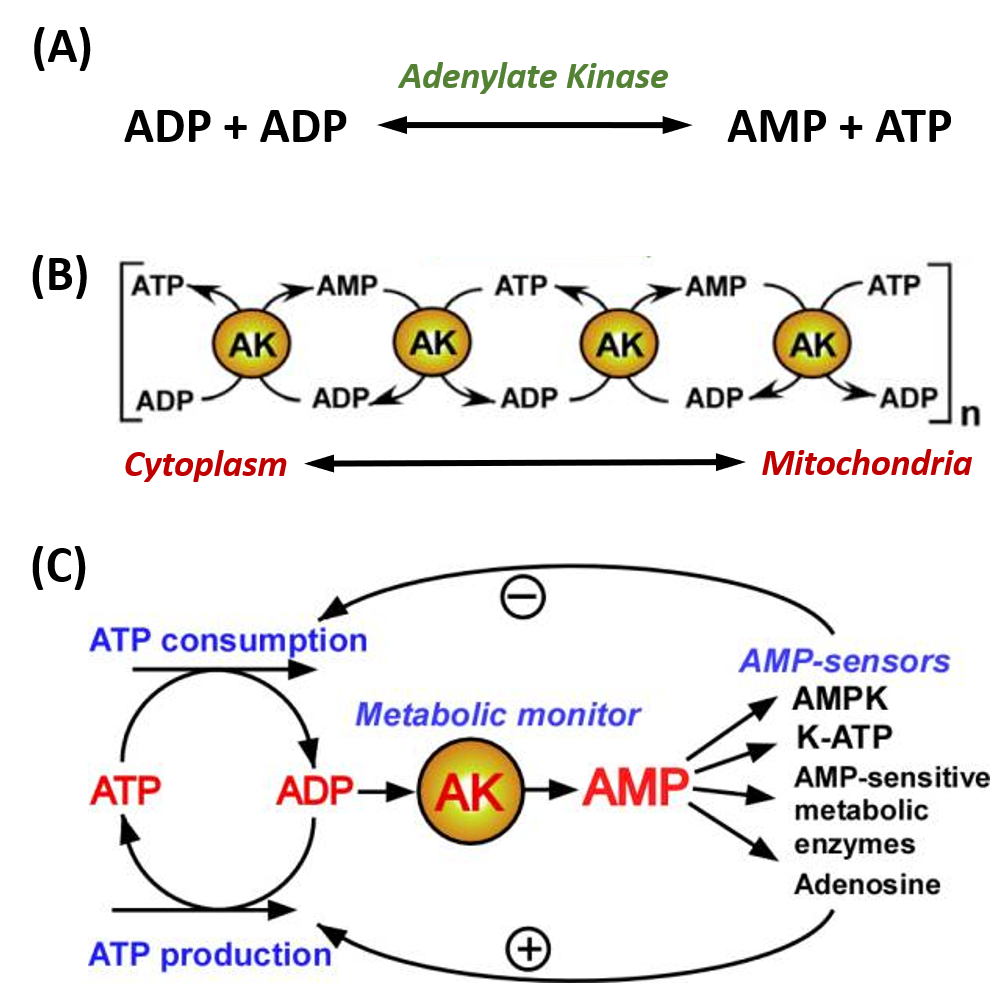
Figure 7.20. Adenylate Kinase (AK) Enzyme Activity. (A) The fundamental reaction of AMP Kinases. (B) reactions of AK enzymes can work in cascading mechanisms to provide signaling and communication between different regions of the cell, including the cytoplasm and mitochondria, and (C) AK enzymes are often used as a metabolic monitor of energy load, leading to the activation or inhibition of downstream enzymes.
Figure modified from: Dzeja, P. and Terzic, A. (2009). Int J Mol Sci 10(4):1729-1772
Phosphoryl transfer during the AK reaction only occurs after the closing of an ‘open lid’ structure in the enzyme through the catalysis by approximation mechanism (Figures 7.21 and 7.22). This causes an exclusion of water molecules that brings the substrates in proximity to each other and effectively lowers the energy barrier for the nucleophilic attack by the γ-phosphoryl group of ATP on the α-phosphoryl of AMP. In the crystal structure of the AK enzyme from E. coli with inhibitor Ap5A (Figure 7.21C), the Arg88 residue coordinate the Ap5A at the α-phosphate group through electrostatic interactions. It has been shown that the mutation of Arg88 to Gly (R88G) results in 99% loss of catalytic activity of this enzyme, suggesting that this residue is intimately involved in the phosphoryl transfer. Another highly conserved residue is Arg119, which lies in the adenosine binding region of the AK, and acts to sandwich the adenine in the active site. It has been suggested that the promiscuity of these enzymes in accepting other NTP’s is due to this relatively inconsequential interactions of the base in the ATP binding pocket. A network of positive, conserved residues (Lys13, Arg123, Arg156, and Arg167 in AK from E. coli) stabilize the buildup of negative charge on phosphoryl group during the transfer. Two distal aspartate residues bind to the arginine network, causing the enzyme to fold and reduces its flexibility. A magnesium cofactor is also required, essential for increasing the electrophilicity of the phosphate on AMP, though this magnesium ion is only held in the active pocket by electrostatic interactions and dissociates easily.
Flexibility and plasticity allow proteins to bind to ligands, form oligomers, aggregate, and perform mechanical work. Large conformational changes in proteins play an important role in cellular signaling. AK acts as a signal transducing protein; thus, the balance between conformations regulates protein activity. AK has an ‘open’ conformation (Figure 7.21A) that is induced into the ‘closed’ and biologically active conformation upon substrate binding.
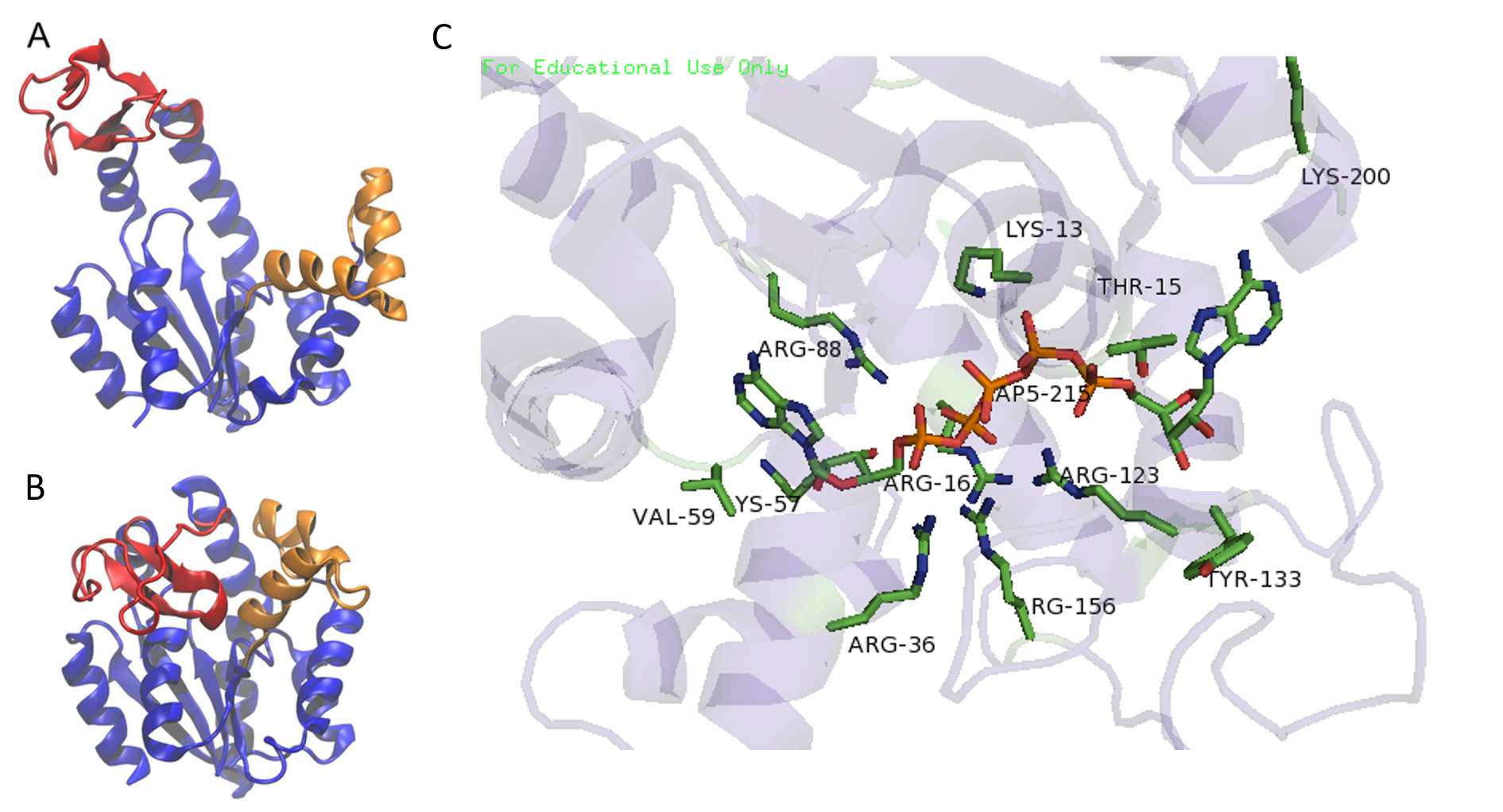
Figure 7.21 Crystal Structure of the Adenylate Kinase Enzyme. Structures of the open (A, PDB ID: 4AKE) and the closed (B, PDB ID: 1AKE) states. The LID and the NMP domains are shown in red and orange respectively. The CORE domain and the rest of the protein are shown in blue. (C) PDB image 3HPQ showing the AK enzyme skeleton in cartoon and the key residues as sticks and labeled according to their placement in the E. coli AK enzyme, crystallized with Ap5A inhibitor.
Figures A and B modified from: Das, A., et al. (2014) PLoS Computational Biology 10(4):e1003521 and Figure C from: Snodgrah
The two subdomains (LID and NMP) can fold and unfold independently of one another depending on substrate binding. Substrate binding induces regional shifts in the protein structure to the partially closed or fully closed conformations. The fully closed conformation optimizes alignment of substrates for phosphoryl-transfer and aids with the removal of water from the active site to avoid wasteful hydrolysis of ATP.
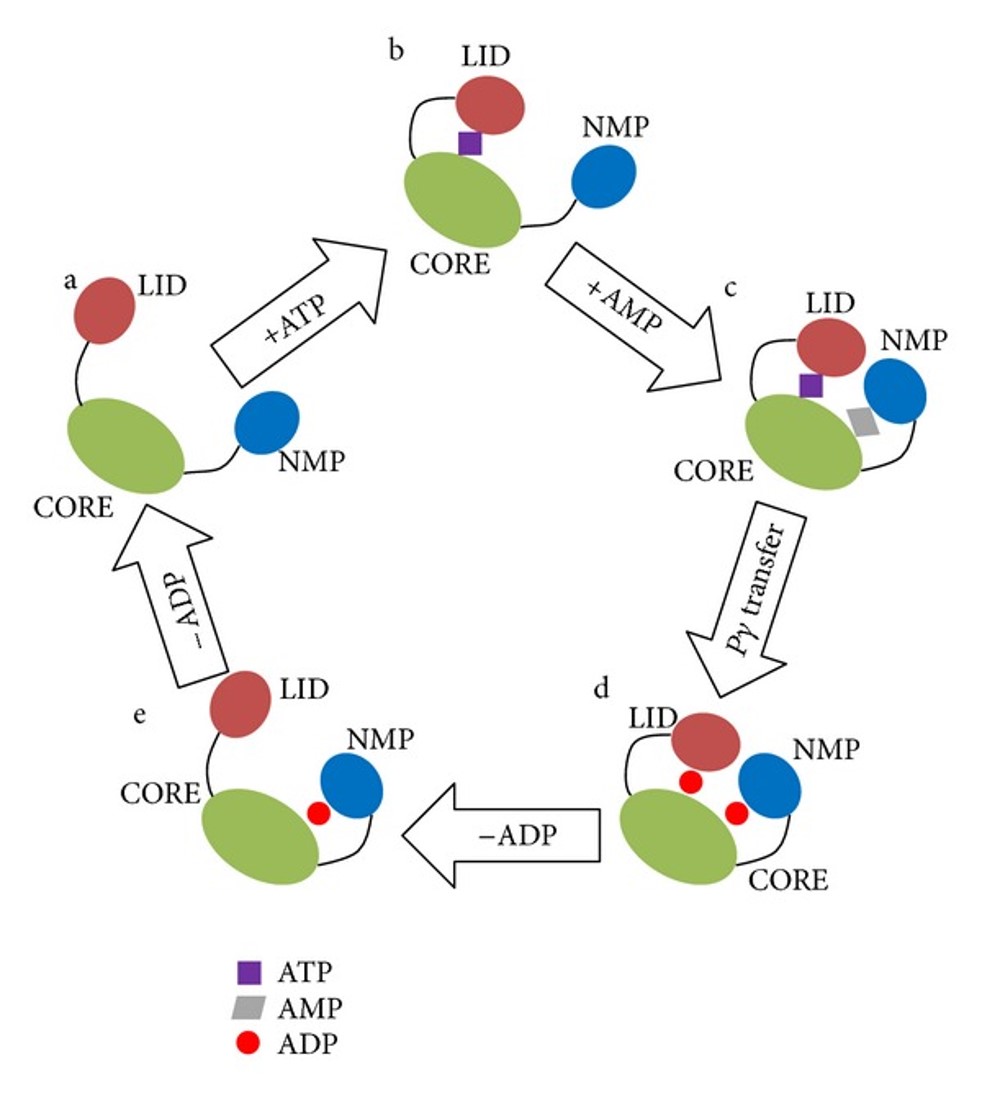
Figure 7.22 Conformational Transition pathway and proposed catalytic mechanism of AK. Model a, substrate free AK with an open conformation. Model b, ATP bound form of ADK with a closed LID domain. Model c, ATP and AMP bound form of AK with a closed conformation. Model d, two ADP bound forms of AK with a closed conformation. Model e, one ADP bound form of AK with a closed NMP domain.
Figure from: Ping, J., et al, (2013) BioMed Res Int: 628536
Restriction Endonucleases
A restriction enzyme, restriction endonuclease, or restrictase is an enzyme that cleaves DNA into fragments at or near specific recognition sites within molecules known as restriction sites. Restriction enzymes are one class of the broader endonuclease group of enzymes. Restriction enzymes are commonly classified into five types, which differ in their structure and whether they cut their DNA substrate at their recognition site, or if the recognition and cleavage sites are separate from one another. To cut DNA, all restriction enzymes make two incisions, once through each sugar-phosphate backbone (i.e. each strand) of the DNA double helix. Here we will focus on the Type II restriction enzymes that are routinely used in molecular biology and biotechnology applications.
As with other classes of restriction enzymes, Type II Restriction Enzymes occur exclusively in unicellular microbial life forms––mainly bacteria and archaea (prokaryotes)––and are thought to function primarily to protect these cells from viruses and other infectious DNA molecules. Inside a prokaryote, the restriction enzymes selectively cut up foreign DNA in a process called restriction digestion; meanwhile, host DNA is protected by a modification enzyme (a methyltransferase) that modifies the prokaryotic DNA and blocks cleavage. Together, these two processes form the restriction modification system.
The first Type II Restriction Enzyme discovered was HindII from the bacterium Haemophilus influenzae Rd. The event was described by Hamilton Smith (Figure 7.23) in his Nobel lecture, delivered on 8 December 1978:
‘In one such experiment we happened to use labeled DNA from phage P22, a bacterial virus I had worked with for several years before coming to Hopkins. To our surprise, we could not recover the foreign DNA from the cells. With Meselson’s recent report in our minds, we immediately suspected that it might be undergoing restriction, and our experience with viscometry told us that this would be a good assay for such an activity. The following day, two viscometers were set up, one containing P22 DNA and the other Haemophilus DNA. Cell extract was added to each and we began quickly taking measurements. As the experiment progressed, we became increasingly excited as the viscosity of the Haemophilus DNA held steady while the P22 DNA viscosity fell. We were confident that we had discovered a new and highly active restriction enzyme. Furthermore, it appeared to require only Mg2+ as a cofactor, suggesting that it would prove to be a simpler enzyme than that from E. coli K or B.
After several false starts and many tedious hours with our laborious, but sensitive viscometer assay, Wilcox and I succeeded in obtaining a purified preparation of the restriction enzyme. We next used sucrose gradient centrifugation to show that the purified enzyme selectively degraded duplex, but not single-stranded, P22 DNA to fragments averaging around 100 bp in length, while Haemophilus DNA present in the same reaction mixture was untouched. No free nucleotides were released during the reaction, nor could we detect any nicks in the DNA products. Thus, the enzyme was clearly an endonuclease that produced double-strand breaks and was specific for foreign DNA. Since the final (limit) digestion products of foreign DNA remained large, it seemed to us that cleavage must be site-specific. This proved to be case and we were able to demonstrate it directly by sequencing the termini of the cleavage fragments.’

Figure 7.23. Hamilton Smith and Daniel Nathans at the Nobel Prize press conference, 12 October 1978 (reproduced with permission from Susie Fitzhugh). Original Repository: Alan Mason Chesney Medical Archives, Daniel Nathans Collection.
Image from: Pingoud, A., Wilson, G.G., and Wende, W. (2014) Nuc Acids Res 42(12):7489-7527.
Restriction enzymes are named according to the taxonomy of the organism in which they were discovered. The first letter of the enzyme refers to the genus of the organism and the second and third to the species. This is followed by letters and/or numbers identifying the isolate. Roman numerals are used to specify different enzymes from the same organism. For example, the enzyme ‘HindIII’ was discovered in Haemophilus influenzae, serotype d, and is distinct from the HindI and HindII endonucleases also present within this bacterium. The DNA-methyltransferases (MTases) that accompany restriction enzymes are named in the same way, and given the prefix ‘M.’. When there is more than one MTase, they are prefixed ‘M1.’, ‘M2.’, etc, if they are separate proteins or ‘M1∼M2.’ when they are joined.
Restriction Enzymes that recognize the same DNA sequence, regardless of where they cut, are termed ‘isoschizomers’ (iso = equal; skhizo = split). Isoschizomers that cut the same sequence at different positions are further termed ‘neoschizomers’ (neo = new). Isoschizomers that cut at the same position are frequently, but not always, evolutionarily drifted versions of the same enzyme (e.g. BamHI and OkrAI). Neoschizomers, on the other hand, are often evolutionarily unrelated enzymes (e.g.EcoRII and MvaI).
Type II Restriction Enzymes are a conglomeration of many different proteins that, by definition, have the common ability to cleave duplex DNA at a fixed position within, or close to, their recognition sequence. This cleavage generates reproducible DNA fragments, and predictable gel electrophoresis patterns, properties that have made these enzymes invaluable reagents for laboratory DNA manipulation and investigation. Almost all Type II Restriction Enzymes require divalent cations, usually Mg2+, as essential components of their catalytic sites. Ca2+, on the other hand, often acts as an inhibitor of Type II Restriction Enzymes.
The recognition sequences of Type II Restriction Enzymes are palindromic in nature, with two possible types of palindromic sequences. The mirror-like palindrome is similar to those found in ordinary text, in which a sequence reads the same forward and backward on a single strand of DNA, as in GTAATG. The inverted repeat palindrome is also a sequence that reads the same forward and backward, but the forward and backward sequences are found in complementary DNA strands (i.e., of double-stranded DNA), as in GTATAC (GTATAC being complementary to CATATG). Inverted repeat palindromes are more common and have greater biological importance than mirror-like palindromes. The position of cleavage within the palindromic sequence can vary depending on the enzyme and can produce either single stranded overhanging sequences (sticky ends) or blunt-ended DNA products.
The EcoRI restriction enzyme produces sticky ends:
![]()
whereas SmaI restriction enzyme cleavage produces blunt-ends:
![]()
Methylation can be used by the host to protect its own genome from cleavage. For example, the methylation of the EcoRI recognition sequence by the M.EcoRI methyltransferase (MTase), changes the sequence from GAATTC to GAm6ATTC (m6A = N6-methyladenine). This modification completely protects the sequence from cleavage by EcoRI.
Type II Restriction Enzymes initially bind non-specifically with the DNA and proceed to slide down the DNA scanning for recognition sequences (Fig 7.24). Upon binding to the correct palindromic sequence the enzyme associates with the metal cofactor and mediates catalytic cleavage of the DNA using the mechanism of strain distortion and catalysis by approximation.
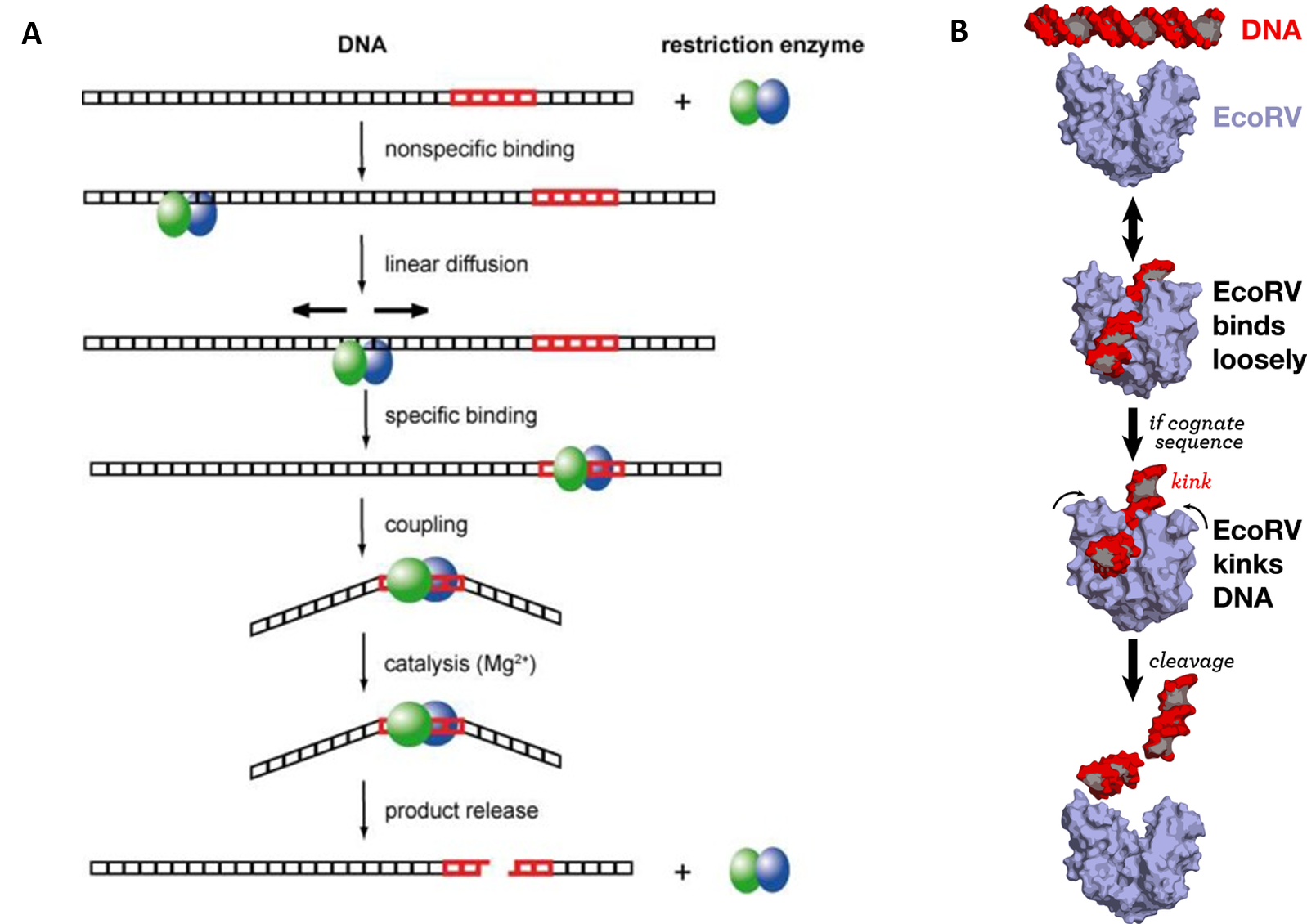
Figure 7.24 DNA Recognition and Cleavage by Type II Restriction Endonucleases. (A) Pictorial view of an EcoRV dimer scanning nonspecifically along the DNA until a specific binding site is recognized. This causes coupling with the metal cofactor and strain distortion of the DNA. Hydrolysis of the phosphodiester bond is mediated and the DNA cleavage products released from the enzyme. (B) shows a space filling model of EcoRV DNA recognition and cleavage.
Figure (A) from: Pingoud, A., Wilson, G.G., and Wende, W. (2014) Nuc Acids Res 42(12):7489-7527. and Figure (B) from: Thomas Splettstoesser
One of the most important questions regarding the catalytic mechanism of a hydrolase is whether hydrolysis involves a covalent intermediate, as is typical for the proteases described previously. This can be decided by analyzing the stereochemical course of the reaction. This was done first for EcoRI, and later for EcoRV. Both enzymes were found to cleave the phosphodiester bond with inversion of stereoconfiguration at the phosphorus, which argues against the formation of a covalent enzyme–DNA intermediate. Thus, it is proposed that cleavage involves the direct nucleophilic attack of the substrate by a water molecule, as shown in Figure 7.25.
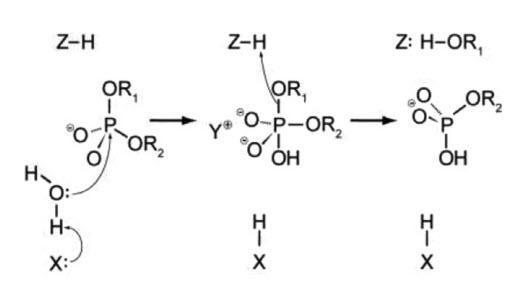
Figure 7.25 A General Mechanism for DNA Cleavage by EcoRI and EcoRV. An activated water molecule attacks the phosphorous in-line wiht the phosphodiester bond to be cleaved, which proceeds with inversion of configuration. X, Y, and Z are a general base, a Lewis acid and a general acid, respectively.
Figure from: Pingoud, A., Wilson, G.G., and Wende, W. (2014) Nuc Acids Res 42(12):7489-7527.
Type II restriction enzymes typically form a homodimer when binding with DNA, as shown in the crystal structure of BglII in Figure 7.26B. BglII catalyses phosphodiester bond cleavage at the DNA backbone through a phosphoryl transfer to water. Studies on the mechanism of restriction enzymes have revealed several general features that seem to be true in almost all cases, although the actual mechanism for each enzyme is most likely some variation of this general mechanism (Figure 7.25). This mechanism requires a base to generate the hydroxide ion from water, which will act as the nucleophile and attack the phosphorus in the phosphodiester bond. Also required is a Lewis acid to stabilize the extra negative charge of the pentacoordinated transition state phosphorus, as well as a general acid or metal ion that stabilizes the leaving group (3’-O−). In some Type II Restriction Enzymes, two divalent metal cofactors are required (such as in EcoRV and BamHI), whereas other enzymes only require one divalent metal cofactor (such as in EcoRI and BglII).
Structural studies of endonucleases have revealed a similar architecture for the active site with the residues following the weak consensus sequence Glu/Asp-(X)9-20-Glu/Asp/Ser-X-Lys/Glu. BglII’s active site is similar to other endonucleases’, following the sequence Asp-(X)9-Glu-X-Gln. In its active site there sits a divalent metal cation, most likely Mg2+, that interacts with Asp-84, Val-94, a phosphoryl oxygen, and three water molecules. One of these water molecules, is able act as a nucleophile because of its proximity to the scissile phosphoryl group (Figure 7.26A). The nucleophilic water molecule is positioned for attack onto the phosphoryl group by a hydrogen bond with the side chain amide oxygen of Gln-95 and its contact with the metal cation. Interaction with the metal cation effectively lowers its pKa, promoting the water’s nucleophilicity (Figure 7.26 A). During hydrolysis, the divalent cation is able to stabilize the 3′-O– leaving group and coordinate proton abstraction from one of the coordinated water molecules (Figure 7.26A).
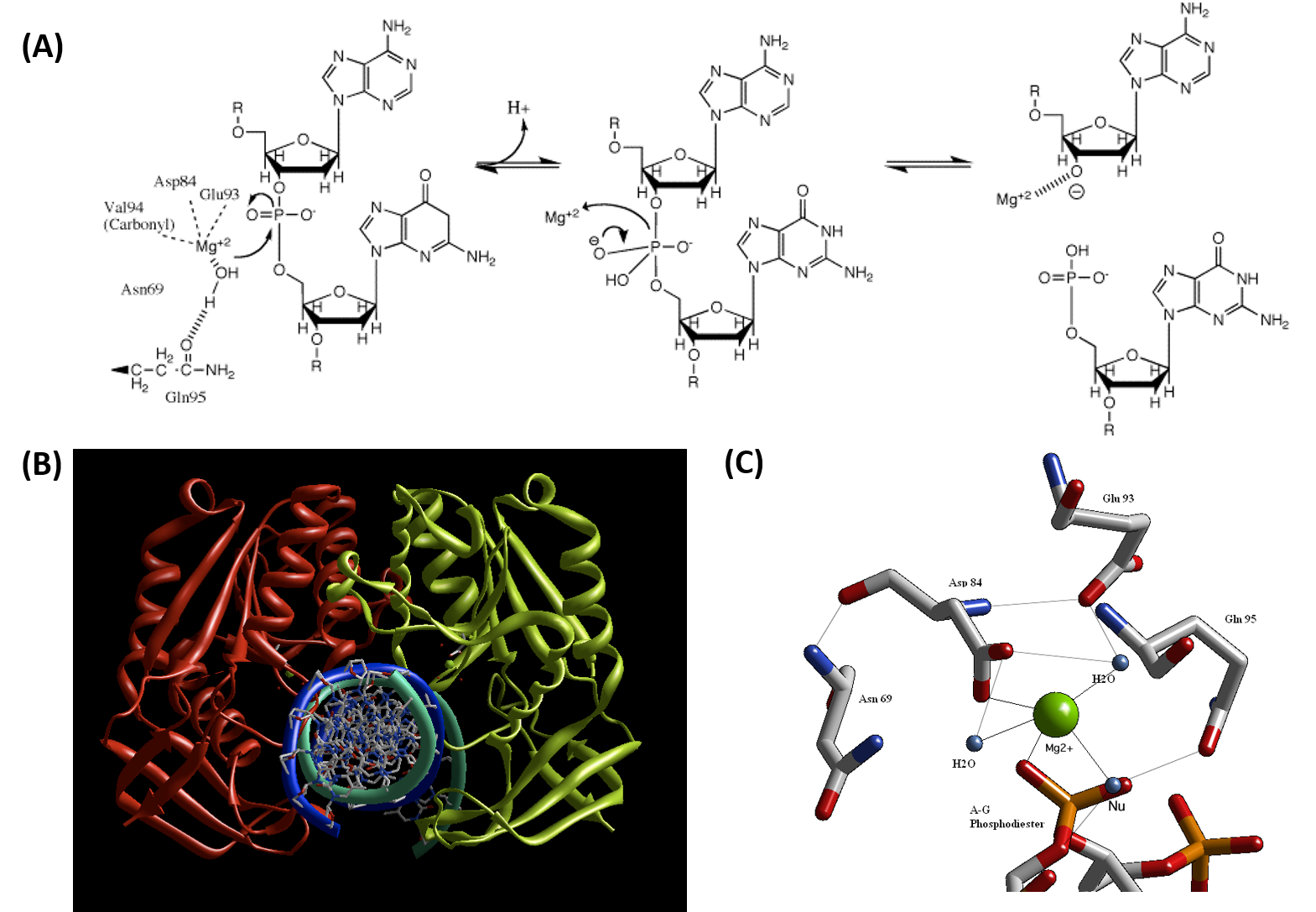
Figure 7.26 Proposed Reaction Mechanism for the Type II Restriction Endonuclease, BglII. (A) Schematic diagram of the catalytic mechanism demonstrating the utility of Mg2+ ions and polar amino acid residues within the active site to activate and position a water molecule for nucleophilic attack on the phosphodiester bond of the DNA substrate. (B) Crystal structure of the BglII dimer with double stranded DNA and (C) Coordination of the Mg2+ cofactor within the active site of the BglII enzyme.
Figures from: GWilliams
Ribozymes
Ribozymes (ribonucleic acid enzymes) are RNA molecules that have the ability to catalyze specific biochemical reactions, including RNA splicing in gene expression, similar to the action of protein enzymes. In 1982, the self-splicing Group I intron was reported as the first discovered catalytic RNA. It was described in the ciliate protozoa Tetrahymena thermofila by Sidney Altman and Thomas Czech, who were awarded the Nobel Prize in Chemistry in 1989. Similar introns can be found in some prokaryotic genomes and the mitochondria and chloroplast DNA of diverse eukaryotes. The second example of a ribozyme to be discovered was the RNAse P involved in tRNA maturation, which had a key biological role and a ubiquitous occurrence in both prokaryotes and eukaryotes. The third reported catalytic RNA was a tiny ribozyme (~50 nt), the self-cleaving hammerhead ribozyme (HHR), which was found in a group of atypical plant pathogens with small circular RNA (circRNA) genomes such as viral satellite RNAs and viroids. Since then, a few more examples of either natural or artificial ribozymes have been discovered, including the ribosome, a singular catalytic RNA that catalyzes the peptide bond formation, the central chemical reaction in extant biology. This landscape strongly supports the hypothesis of a prebiotic RNA world, where the first self-replicating organisms were based on RNA as both the genetic material and as catalyst. Whereas modern proteins would have replaced most of these ancient catalytic RNAs, some of them have remained in current organisms performing different functions. Among all known ribozymes, there is the enigmatic family of small (<200 nt) self-cleaving RNAs, which catalyse a simple intramolecular transesterification in a highly sequence-specific manner. This reaction, which can occur spontaneously in the RNA, starts by a SN2-like nucleophilic attack of the 2′-oxygen to the adjacent 3′-phosphate, resulting in cleavage of the phosphodiester bond to form a 2′-3′-cyclic phosphate and a 5′-hydroxyl RNA products (Figure 7.27A).
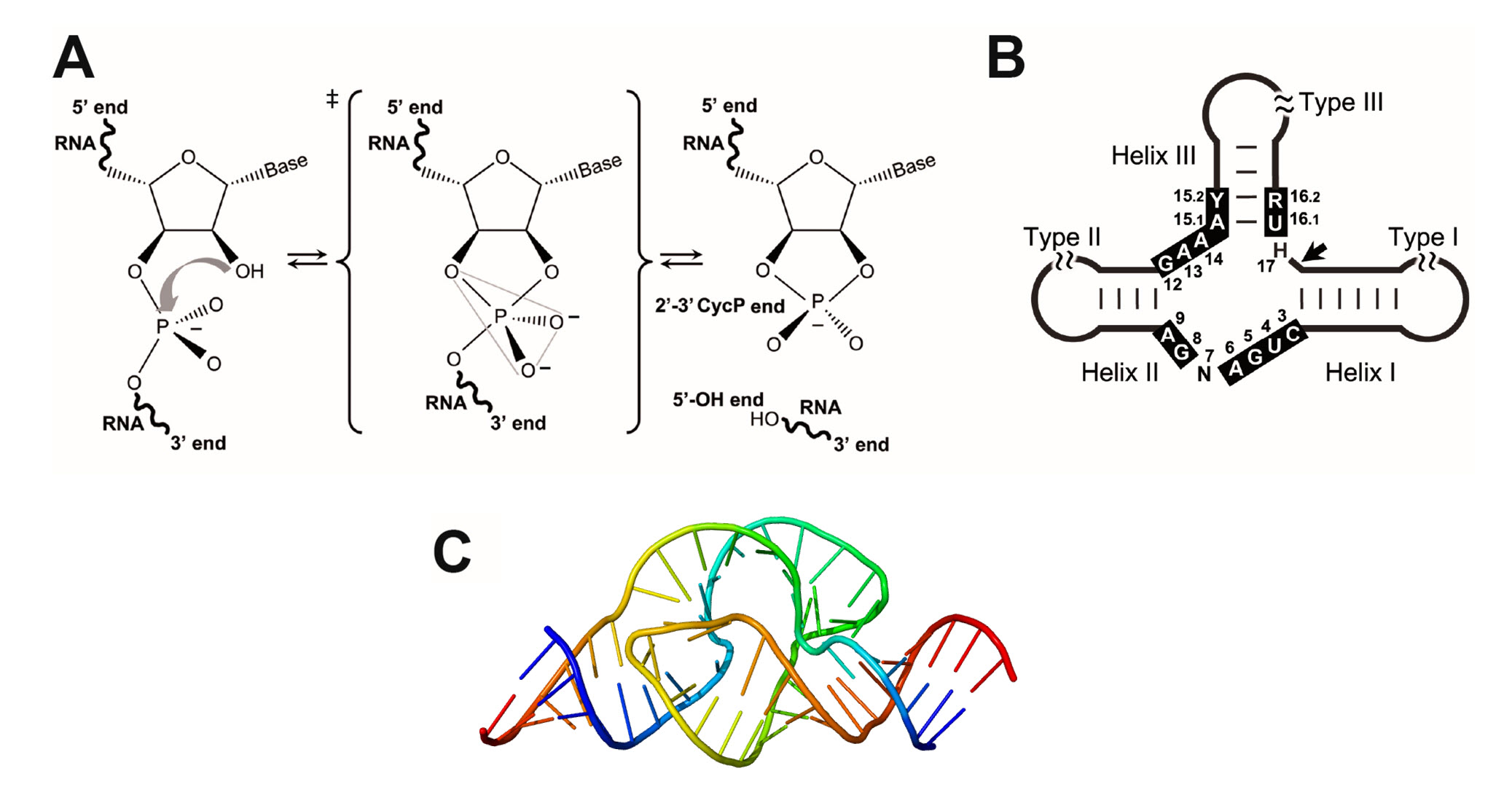
Figure 7.27 The Hammerhead Ribozyme. (A) Mechanism of internal transesterification reaction in the RNA. The cleavage reaction proceeds with an attack of the hydroxyl moiety at 2′ to the phosphate group at 3′, followed by a bipyramidal transition-state. The cleavage products are a 2′-3′-cyclic phosphate at the 5′ RNA product and a 5′-hydroxyl at the 3′ RNA product; (B) Diagram of the hammerhead ribozyme. Black boxes indicate the highly conserved nucleotides at the catalytic core. Secondary structures of (C) 3-Dimensional structure of the Hammerhead Ribozyme.
Figures A and B from: De la Peña, M., et al (2017) Molecules 22(1):78 and Figure C from: Wgscott
Similar to ribonuclease proteins such as the RNAse A, small self-cleaving ribozymes stabilize the formation of the bipyramidal oxyphosphorane transition-state through different catalytic strategies, such as in-line atomic orientation, electrostatic neutralization, and general acid-base catalysis. In this way, nucleolytic ribozymes are able to catalyze RNA cleavage at a rate only a few-fold slower than their protein counterparts. At least nine classes of naturally-occurring small self-cleaving ribozymes have been described so far: the hammerhead (Figure 7.27C), hairpin, human Hepatitis-δ, Varkud-satellite, GlmS, twister, twister sister, hatchet and pistol ribozymes. Since its discovery 30 years ago, the HHR has been extensively used as a model ribozyme for structural, biochemical and biological studies. It is composed of a catalytic centre comprising 15 highly conserved nucleotides surrounded by three double helixes (I to III), which adopt a secondary structure that resembles the shape of a hammerhead shark head. Depending on the open-ended helix, there are three possible circularly permuted forms, named type I, II or III (Figure 7.27B). The HHR motif, like other small ribozymes such as hairpin and Hepatitis-δ, has been historically regarded as a biological peculiarity of subviral circular RNA genomes. However, we know now that small catalytic RNAs such as the HHR can occur numerously in DNA genomes from bacteria to eukaryotes, including our own genome, and carry out diverse biological functions that we are just starting to recognize.
The ribosome also functions as a ribozyme mediating the formation of the amide bind during protein synthesis. Protein synthesis from a mRNA template occurs on a ribosome, a nanomachine composed of proteins and ribosomal RNAs (rRNA). The ribosome is composed of two very large structural units that are an amalgamation of proteins and rRNA molecules (Figure 7.28). The smaller unit (termed 30S and 40S in bacteria and eukaryotes, respectively) coordinates the correct base pairing of the triplet codon on the mRNA with another small adapter RNA, transfer or tRNA, that brings a covalently connected amino acid to the site.

Figure 7.28 The Small Ribosomal Subunit of Thermus thermophilus. The 16S ribosomal RNA is shown in orange with ribosomal proteins attached in blue.
Figure from: Goodsell, D.S. – Molecule of the Month
Peptide bond formation occurs when another tRNA-amino acid molecule binds to an adjacent codon on mRNA. The tRNA has a cloverleaf tertiary structure with some intrastranded H-bonded secondary structure. The last three nucleotides at the 3′ end of the tRNA are CpCpA. The amino acid is esterified to the terminal 3’OH of the terminal A by a protein enzyme, aminoacyl-tRNA synthetase.
Covalent amide bond formation between the second amino acid to the first, forming a dipeptide, occurs at the peptidyl transferase center, located on the larger ribosomal subunit (50S and 60S in bacteria and eukaryotes, respectively). The ribosome ratchets down the mRNA so the dipeptide-tRNA is now at the the P or Peptide site, awaiting a new tRNA-amino acid at the A or Amino site. Figure 7.29 shows a schematic of the ribosome with bound mRNA on the 30S subunit and tRNAs covalently attached to amino acid (or the growing peptide) at the A and P site, respectively.
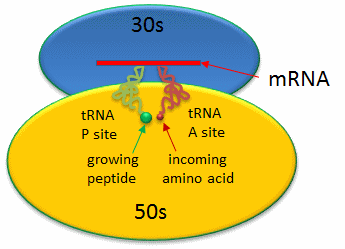
Figure 7.29 Schematic Representation of a Bacterial Ribosome. The 50s (yellow) and 30s (blue) subunits of the ribosome are composed of protein and rRNA. mRNA (red linear strand) is shown docked onto the 30s subunit. The P and A sites are filled with tRNA molecules (green and red).
Figure from: Jakubowski, H.
A likely mechanism for the formation of the amide bond between a growing peptide on the P-site tRNA and the amino acid on the A-site tRNA has been derived from crystal structures with bound substrates and transition state analogs and is shown in Figure 7.30. Catalysis does not involve any of the ribosomal proteins (not shown) since none is close enough to the peptidyl transferase center to provide amino acids that could participate in general acid/base catalysis. Hence the rRNA must acts as the enzyme (i.e. it is a ribozyme). Initially it was thought that a proximal adenosine with a perturbed pKa could, at physiological pH, be protonated/deprotonated and hence act as a general acid/base in the reaction. However, none was found. The most likely mechanism to stabilize the oxyanion transition state at the electrophilic carbon attack site is precisely located water, which is positioned at the oxyanion hole by H-bonds to uracil 2584 on the rRNA. The cleavage mechanism involves the concerted proton shuffle shown Figure 7.30. In this mechanism, the substrate (Peptide-tRNA) assists its own cleavage in that the 2’OH is in position to initiate the protein shuttle mechanism. (A similar mechanism might occur to facilitate hydrolysis of the fully elongated protein from the P-site tRNA.) Of course all of this requires perfect positioning of the substrates and isn’t that what enzymes do best? The main mechanisms for catalysis of peptide bond formation by the ribosome (as a ribozyme) are intramolecular catalysis and transition state stabilization by the appropriately positioned water molecule.
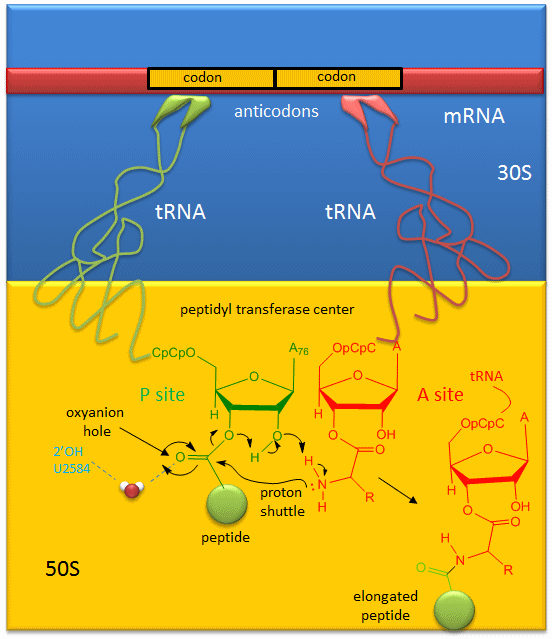
Figure 7.30 Mechanism of Peptide Bond Formation. Peptide bond formation is likely mediated by a proton shuttle mechanism that is stabilized by a coordinated water molecule within the oxyanion hole.
Figure from: Jakubowski, H.
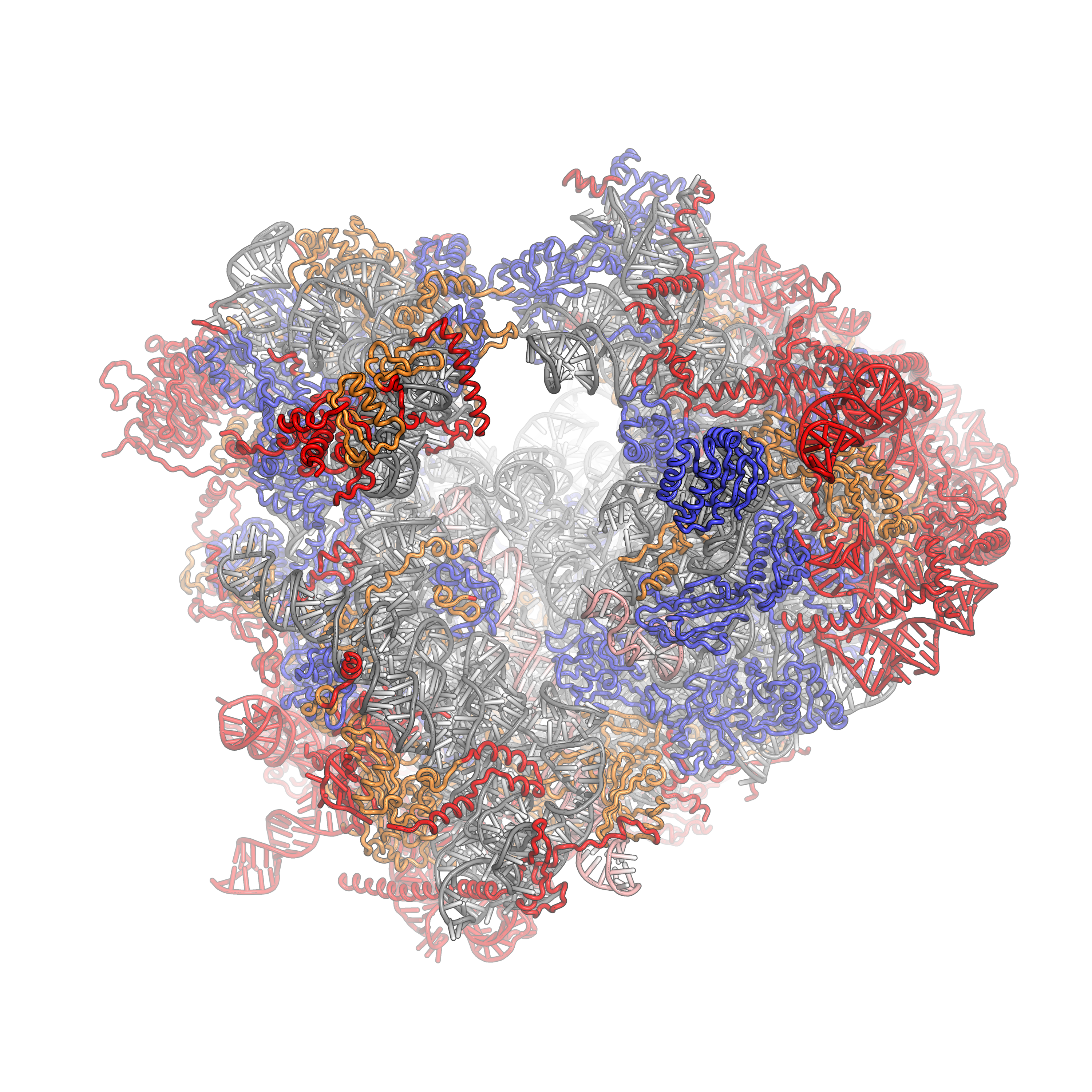
The crystal structure of the eukaryotic ribosome has recently been published (Ben-Shem et al). It is significantly larger (40%) than the prokaryotic counterpart, with mass of around 3×106 Daltons. The 40S subunit has one rRNA chain (18S) and 33 associated proteins, while the larger 60S subunit has 3 rRNA chains (25S, 5.8S and 5S) and 46 associated proteins. The larger size of the eukaryotic ribosome facilitates more interactions with cellular proteins and greater regulation of cellular events. The structure of a eukaryotic 80S ribosome showing rRNA and protein interactions is shown in Figure 7.31.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 7.31 Eukaryotic 80S Ribosome. The 40S subunit is on the left, the 60S subunit on the right. The ribosomal RNA (rRNA) core is represented as a grey tube, expansion segments are shown in red. Universally conserved proteins are shown in blue. These proteins have homologs in eukaryotes, archaea and bacteria. Proteins shared only between eukaryotes and archaea are shown in orange, and proteins specific to eukaryotes are shown in red. PDB identifiers 4a17, 4A19, 2XZM aligned to 3U5B, 3U5C, 3U5D, 3U5E
Figure from: Fvoigtsh
#/media/File:80S_2XZM_4A17_4A19.png){kind=link}
7.4 References:
- Wikipedia contributors. (2020, April 21). Nucleophile. In Wikipedia, The Free Encyclopedia. Retrieved 15:39, April 26, 2020, from https://en.wikipedia.org/w/index.php?title=Nucleophile&oldid=952368939
- Oregon Institute of Technology (2019) Organic Chemistry II (Lund). In Libretexts. Retrieved 10:58 am, April 27, 2020 from: https://chem.libretexts.org/Courses/Oregon_Institute_of_Technology/OIT%3A_CHE_332_–_Organic_Chemistry_II_(Lund)
- Wikipedia contributors. (2020, April 12). Bond cleavage. In Wikipedia, The Free Encyclopedia. Retrieved 15:15, April 27, 2020, from https://en.wikipedia.org/w/index.php?title=Bond_cleavage&oldid=950494652
- Wikipedia contributors. (2020, February 24). Arrow pushing. In Wikipedia, The Free Encyclopedia. Retrieved 15:25, April 27, 2020, from https://en.wikipedia.org/w/index.php?title=Arrow_pushing&oldid=942438883
- Wikipedia contributors. (2020, April 16). Acid dissociation constant. In Wikipedia, The Free Encyclopedia. Retrieved 15:48, April 27, 2020, from https://en.wikipedia.org/w/index.phptitle=Acid_dissociation_constant&oldid=951313744
- Farmer, S., Reusch, W., Alexander, E., and Rahim, A. (2016) Organic Chemistry. Libretexts. Available at: https://chem.libretexts.org/Core/Organic_Chemistry
- Ball, et al. (2016) MAP: The Basics of GOB Chemistry. Libretexts. Available at: https://chem.libretexts.org/Textbook_Maps/Introductory_Chemistry_Textbook_Maps/Map%3A_The_Basics_of_GOB_Chemistry_(Ball_et_al.)/14%3A_Organic_Compounds_of_Oxygen/14.10%3A_Properties_of_Aldehydes_and_Ketones
- McMurray (2017) MAP: Organic Chemistry. Libretexts. Available at: https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry_Textbook_Maps/Map%3A_Organic_Chemistry_(McMurry)
- Soderburg (2015) Map: Organic Chemistry with a Biological Emphasis. Libretexts. Available at: https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry_Textbook_Maps/Map%3A_Organic_Chemistry_With_a_Biological_Emphasis_(Soderberg)
- Ophardt, C. (2013) Biological Chemistry. Libretexts. Available at: https://chem.libretexts.org/Core/Biological_Chemistry/Proteins/Case_Studies%3A_Proteins/Permanent_Hair_Wave
- Soderberg, T. (2016) Organic Chemistry with a Biological Emphasis. Libretexts. Available at: https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry_Textbook_Maps/Map%3A_Organic_Chemistry_with_a_Biological_Emphasis_(Soderberg)
- Ball, et al. (2016) MAP: The Basics of General, Organic, and Biological Chemistry. Libretexts. Available at: https://chem.libretexts.org/Textbook_Maps/Introductory_Chemistry_Textbook_Maps/Map%3A_The_Basics_of_GOB_Chemistry_(Ball_et_al.)
- Clark, J. (2017) Organic Chemistry. Libretexts. Available at: https://chem.libretexts.org/Core/Organic_Chemistry/Amides/Reactivity_of_Amides/Polyamides
- Wikipedia contributors. (2018, December 28). Metabolism. In Wikipedia, The Free Encyclopedia. Retrieved 19:28, December 29, 2018, from https://en.wikipedia.org/w/index.php?title=Metabolism&oldid=875751739
- Ball, Hill, and Scott. (2012) Enzyme Activity, section 18.7 from the book Introduction to Chemistry: General, Organic and Biological (v1.0) retrieved on Dec 31, 2018 from https://2012books.lardbucket.org/books/introduction-to-chemistry-general-organic-and-biological/s21-07-enzyme-activity.html
- Wikipedia contributors. (2018, November 29). Mechanism of action. In Wikipedia, The Free Encyclopedia. Retrieved 05:00, January 1, 2019, from https://en.wikipedia.org/w/index.php?title=Mechanism_of_action&oldid=871201209
- Mótyán, J.A., Tóth, F., and Tőzsér, J. (2013) Research Applications of Proteolytic Enzymes in Molecular Biology. Biomolecules 3(4), 923-942; https://doi.org/10.3390/biom3040923
- Wikipedia contributors. (2020, April 11). Adenylate kinase. In Wikipedia, The Free Encyclopedia. Retrieved 19:28, May 4, 2020, from https://en.wikipedia.org/w/index.php?title=Adenylate_kinase&oldid=950311736
- Ahern, K., Rajagopal, I., and Tan, T. (2019) Biochemistry Free and Easy. Available at Oregon State University (http://biochem.science.oregonstate.edu/content/biochemistry-free-and-easy) and Libretexts (https://bio.libretexts.org/Bookshelves/Biochemistry/Book%3A_Biochemistry_Free_For_All_(Ahern%2C_Rajagopal%2C_and_Tan)/04%3A_Catalysis/4.03%3A_Mechanisms_of_Catalysis)
- Wikipedia contributors. (2020, April 16). Serine protease. In Wikipedia, The Free Encyclopedia. Retrieved 14:32, May 6, 2020, from https://en.wikipedia.org/w/index.php?title=Serine_protease&oldid=951309456
- Wikipedia contributors. (2020, April 16). Restriction enzyme. In Wikipedia, The Free Encyclopedia. Retrieved 15:12, May 16, 2020, from https://en.wikipedia.org/w/index.php?title=Restriction_enzyme&oldid=951351229
- Pingoud, A., Wilson, G.G., and Wende, W. (2014) Type II restriction endonucleases – a historical perspective and more. Nuc Acids Res 42(12)7489-7527. Retrieved from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4081073/pdf/gku447.pdf
- Wikipedia contributors. (2019, July 25). BglII. In Wikipedia, The Free Encyclopedia. Retrieved 20:48, May 16, 2020, from https://en.wikipedia.org/w/index.php?title=BglII&oldid=907885716
- De la Peña, M, GarcÍa-Robles, I., and Cervera, A. (2017) The Hammerhead Ribozyme: A Long History for a Short RNA. Molecules 22(1):78. Retrieved from: https://www.mdpi.com/1420-3049/22/1/78/htm
- Jakubowski, H. (2019) Biochemistry Online. Libretexts. Available at: https://bio.libretexts.org/Bookshelves/Biochemistry/Book%3A_Biochemistry_Online_(Jakubowski)