Home » Student Resources » Online Chemistry Textbooks » CH450 and CH451: Biochemistry - Defining Life at the Molecular Level » Chapter 10: Transcription and RNA Processing
MenuCH450 and CH451: Biochemistry - Defining Life at the Molecular Level
Chapter 10: Transcription and RNA Processing
10.1 Types of RNA
10.2 RNA Polymerase Enzymes
10.3 Transcription Factors and the Preinitiation Complex (PIC)
10.4 Transcriptional Elongation and Termination
10.5 Processing of RNA
10.6 References
10.1 Types of RNA
Structurally speaking, ribonucleic acid (RNA), is quite similar to DNA. However, whereas DNA molecules are typically long and double stranded, RNA molecules are much shorter and are typically single stranded. A ribonucleotide within the RNA chain contains ribose (the pentose sugar), one of the four nitrogenous bases (A, U, G, and C), and a phosphate group. The subtle structural difference between the sugars gives DNA added stability, making DNA more suitable for storage of genetic information, whereas the relative instability of RNA makes it more suitable for its more short-term functions. The RNA-specific pyrimidine uracil forms a complementary base pair with adenine and is used instead of the thymine that is found in DNA. Even though RNA is single stranded, most types of RNA molecules show extensive intramolecular base pairing between complementary sequences within the RNA strand, creating a predictable three-dimensional structure essential for their function (Figures 10.1 and 10.2).

Figure 10.1 RNA Structural Elements (a) Ribonucleotides contain the pentose sugar ribose instead of the deoxyribose found in deoxyribonucleotides. (b) RNA contains the pyrimidine uracil in place of thymine found in DNA.
Figure from:Parker, et al (2019) Microbiology from Openstax
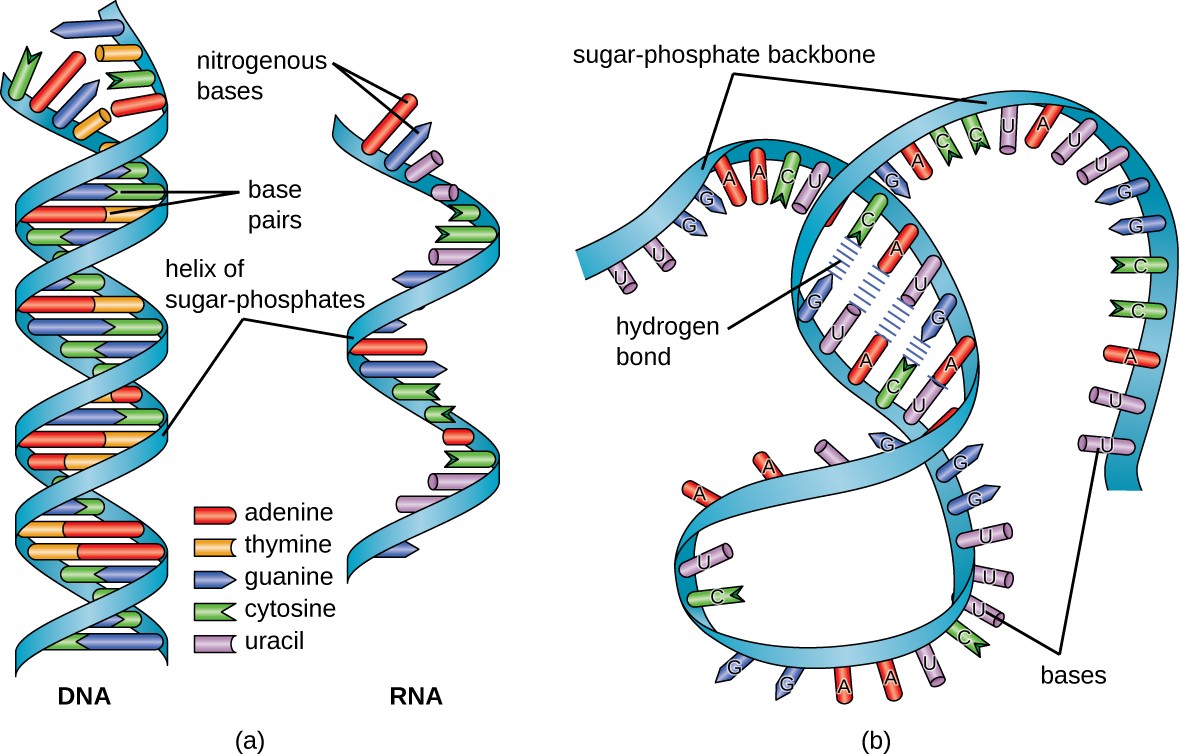
Figure 10.2 Structural Comparison of DNA and RNA (a) DNA is typically double stranded, whereas RNA is typically single stranded. (b) Although it is single stranded, RNA can fold upon itself, with the folds stabilized by short areas of complementary base pairing within the molecule, forming a three-dimensional structure.
Figure from: Parker, et al (2019) Microbiology from Openstax
RNA can largely be divided into two types, one that carries the code for making proteins or coding RNA, which is also called messenger RNA (mRNA), and non-coding RNA (ncRNA). The ncRNA can be subdivided into several different types, depending either on the length of the RNA or on the function. Size classification begins with the short ncRNAs (~20–30 nt), which include microRNAs (miRs), and small interfering (siRNAs); the small ncRNAs up to 200 nt, which include transfer RNA (tRNA), small nuclear RNA (snRNA), and small nucleolar RNA (snoRNA); and long ncRNAs ( > 200 nt), which include ribosomal RNA (rRNA), enhancer RNA (eRNA) and long intergeneic ncRNAs (lincRNAs), among others.
Cells access the information stored in DNA by creating RNA, through the process of transcription, which then directs the synthesis of proteins through the process of translation. The three main types of RNA directly involved in protein synthesis are messenger RNA (mRNA), ribosomal RNA (rRNA), and transfer RNA (tRNA). The mRNA carries the message from the DNA, which controls all of the cellular activities in a cell. If a cell requires a certain protein to be synthesized, the gene for this product is “turned on” and the mRNA is synthesized through the process of transcription. The mRNA then interacts with ribosomes and other cellular machinery to direct the synthesis of the protein it encodes during the process of translation. mRNA is relatively unstable and short-lived in the cell, especially in prokaryotic cells, ensuring that proteins are only made when needed.
rRNA and tRNA are stable types of RNA. In prokaryotes and eukaryotes, tRNA and rRNA are encoded by the DNA, where they are transcribed into long RNA molecules that are subsequently cut to release smaller fragments containing the individual mature RNA species. In eukaryotes, synthesis, cutting, and assembly of rRNA into ribosomes takes place in the nucleolus region of the nucleus, but these activities occur in the cytoplasm of prokaryotes. Within the nucleolus region, ribosome assembly requires the activity of numerous snoRNAs.
Ribosomes are composed of rRNA and protein. As its name suggests, rRNA is a major constituent of ribosomes, composing up to about 60% of the ribosome by mass and providing the location where the mRNA binds. The rRNA ensures the proper alignment of the mRNA, tRNA, and the ribosomes; the rRNA of the ribosome also has an enzymatic activity (peptidyl transferase) and catalyzes the formation of the peptide bonds between two aligned amino acids during protein synthesis (Figure 10.3). Although rRNA had long been thought to serve primarily a structural role, its catalytic role within the ribosome was shown in 2000. Scientists in the laboratories of Thomas Steitz (1940–) and Peter Moore (1939–) at Yale University were able to crystallize the ribosome structure from Haloarcula marismortui, a halophilic archaeon isolated from the Dead Sea. Because of the importance of this work, Steitz shared the 2009 Nobel Prize in Chemistry with other scientists who made significant contributions to the understanding of ribosome structure. The structure and function of ribosomes will be discussed in further detail in Chapter 11.
Transfer RNA (tRNA) is the third prominent type of RNA involved in protein translation. tRNAs are usually only 70–90 nucleotides long. They carry the correct amino acid to the site of protein synthesis in the ribosome. It is the base pairing between the tRNA and mRNA that allows for the correct amino acid to be inserted in the polypeptide chain being synthesized (Figure 10.3). Any mutations in the tRNA or rRNA can result in global problems for the cell because both are necessary for proper protein synthesis.

Figure 10.3. A generalized illustration of how mRNA and tRNA are used in protein synthesis within a cell.
Figure from: Parker, et al (2019) Microbiology from Openstax
As described in Chapter 7, some RNA molecules have enzymatic properties and serve as ribozymes. Within this chapter, the activity of snRNAs during the process of intron removal from mRNA sequences function as ribozymes and will be described. Furthermore, a detailed description of the enzymatic features of the ribosome structure will be provided in Chapter 11.
Other small ncRNA and lncRNA molecules play a role in the regulation of transcriptional and translational processes. For example, the post-transcriptional expression levels of many genes can be controlled by RNA interference, in which miRNAs, specific short RNA molecules, pair with mRNA regions and target them for degradation (Figure 10.4). This process is aided by protein chaperones called argonautes. This antisense-based process involves steps that first process the miRNA so that it can base-pair with a region of its target mRNAs. Once the base pairing occurs, other proteins direct the mRNA to be destroyed by nucleases. Fire and Mello were awarded the 2006 Nobel Prize in Physiology or Medicine for this discovery.
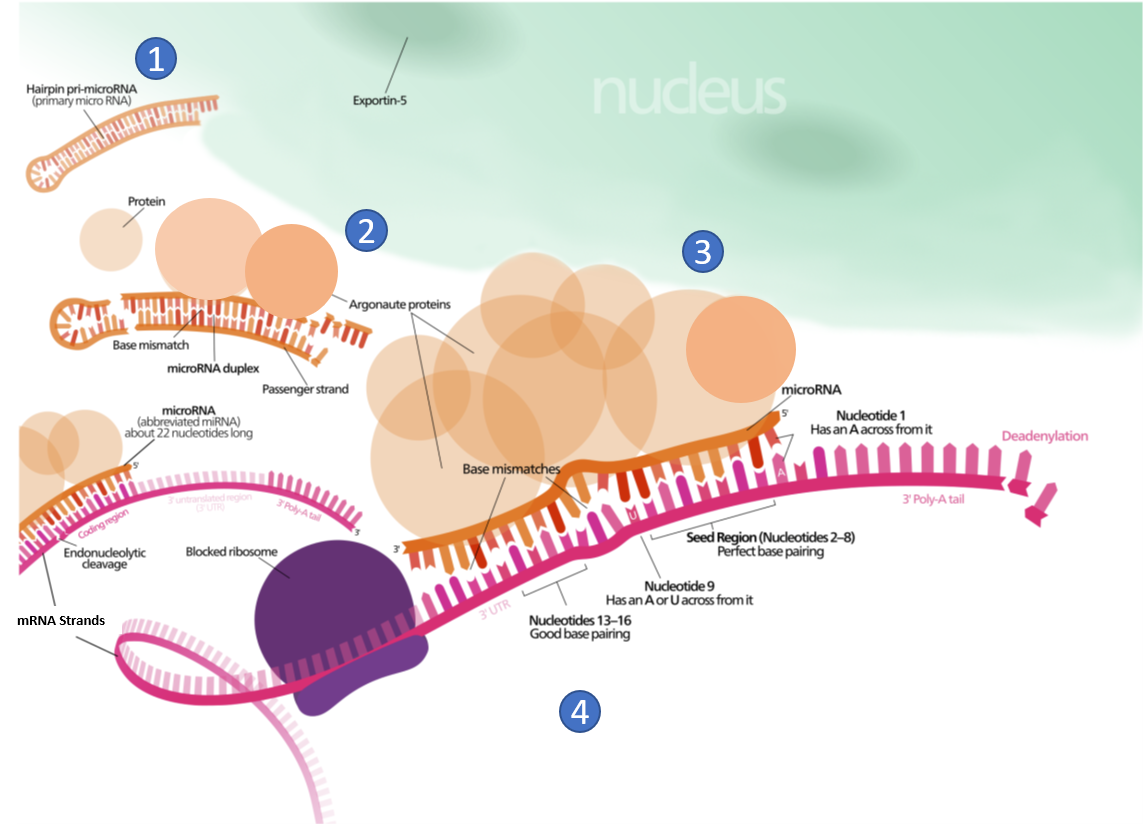
Figure 10.4 Role of Micro RNA (miRNA) in the Inhibition of Eukaryotic mRNA Translation. (1) A protein called Exportin-5 transports a hairpin primary micro RNA (pri-miRNA) out of the nucleus and into the cytoplasm. (2) An enzyme called Dicer (not shown), trims the pri-miRNA and removes the hairpin loop. A group of proteins, known as Argonautes, form a miRNA/protein complex. (3) miRNA/protein complex hydrogen bonds with mRNA based on complimentary sequence homology, and blocks translation. (4) The miRNA/protein complex binding speeds up the breakdown of the polyA tail of the mRNA, causing the mRNA to be degraded sooner.
Figure modified from: Wikimedia Commons
{kind=link}
At steady state, the vast majority of human cellular RNA consists of rRNA (∼90% of total RNA for most cells Figure 10.5). Although there is less tRNA by mass, their small size results in their molar level being higher than rRNA (Figure 10.5). Other abundant RNAs, such as mRNA, snRNA, and snoRNAs are present in aggregate at levels that are about 1–2 orders of magnitude lower than rRNA and tRNA (Figure 10.5). Certain small RNAs, such as miRNA and piRNAs can be present at very high levels; however, this appears to be cell type dependent. lncRNAs are present at levels that are two orders of magnitude less than total mRNA. Although the estimated number of different types of human lncRNAs may have a very restricted expression pattern and thus, accumulate to higher levels within specific cell types. For example, sequencing of mammalian transcriptomes has revealed more than 100,000 different lncRNA molecules can be produced, compared with the approximate 20,000 protein-coding genes. The diversity and functions of the transcriptome within biological processes are currently a highly active area of research.

Figure 10.5: Estimate of RNA levels in a typical mammalian cell. Proportion of the various classes of RNA in mammalian somatic cells by total mass (A) and by absolute number of molecules (B). Total number of RNA molecules is estimated at roughly 107 per cell. Other ncRNAs in (A) include snRNA, snoRNA, and miRNA. Note that due to their relatively large sizes, rRNA, mRNA, and lncRNAs make up a larger proportion of the mass as compared to the overall number of molecules.
Figure from: Palazzo, A. and Lee, E.S. (2015) Frontiers in Genetics 6:2
Back to the Top
10.2 RNA Polymerase Enzymes
RNA Polymerase Enzymes (RNAPs) are required to carry out the process of transcription and are found in all cells ranging from bacteria to humans. All RNAPs are multi-subunit assemblies, with bacteria having five core subunits that have homologs in archaeal and eukaryotic RNAPs. Bacterial RNAPs are the simplest form of RNA polymerases and provide an excellent system to study how they control transcription.
Prokaryotic RNA Polymerase Enzymes
The RNAP catalytic core within bacteria contains five major subunits (α2ββ’ω) (Fig 10.7B). To position this catalytic core onto the correct promoter requires the association of a sixth subunit called the sigma factor (σ). Within bacteria there are multiple different sigma factors that can associate with the catalytic core of RNAP that help to direct the catalytic core to the correct DNA locations where RNAP can then initiate transcription. For example, within E. coli σ70 is the housekeeping sigma factor that is responsible for transcribing most genes in growing cells. It keeps essential genes and pathways operating. Other sigma factors are activated during certain environmental situations, such as σ38 which is activated during starvation or when cells reach the stationary phase. When the sigma subunit associates with the RNAP catalytic core, the RNAP has then formed the holoenzyme. When bound to DNA, the holoenzyme conformation of RNAP can initiate transcription.
Transcription takes place in several stages. To start with, the RNA polymerase holoenzyme locates and binds to promoter DNA. At this stage the RNAP holoenzyme is it the closed conformation (RPc) (Figure 10.6). Initial specific binding to the promoter by sigma factors of the holoenzyme, sets in motion conformational changes in which the RNAP molecular machine bends and wraps the DNA with mobile regions of RNAP playing key roles (Figure 10.6). Next, RNAP separates the two strands of DNA and exposes a portion of the template strand. At this point, the DNA and the holoenzyme are said to be in an ‘open promoter complex’ (RPo), and the section of promoter DNA that is within it is known as a ‘transcription bubble’ (Figure 10.6).
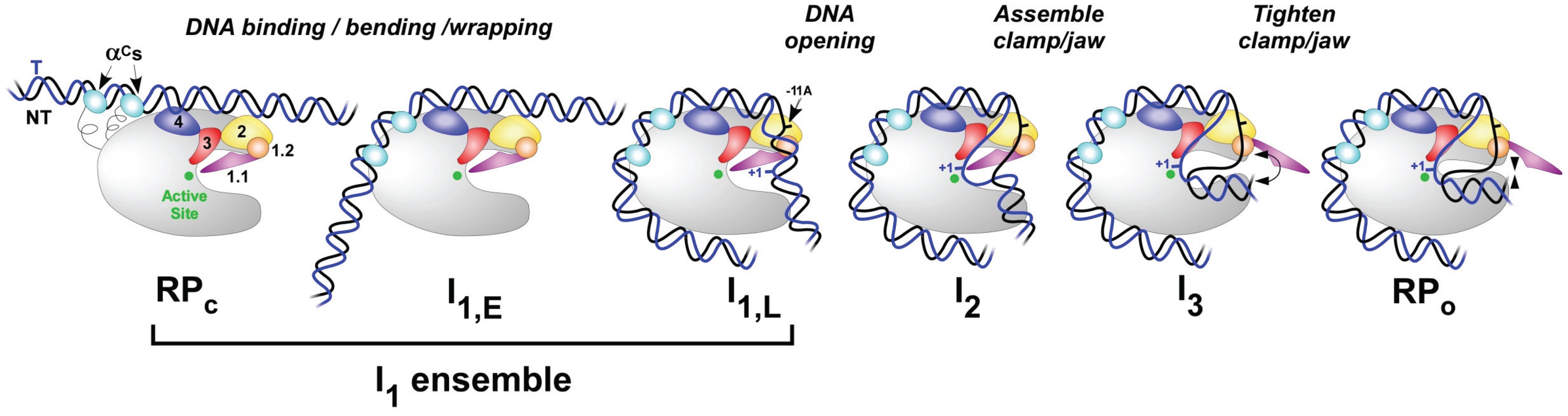
Figure 10.6. Schematic Representation of E.coli Transcriptional Initiation. Closed complexes like RPC, I1,E (early), or I1,L (late) can be significant members of the rapidly equilibrating I1 ensemble. RPo signifies the end of the initiation stage and entry into the elongation phase of RNA synthesis. The α domains are shown in light blue; σ domains are indicated by numbers 1.1, 1.2, 2, 3, and 4.
Figure from: Ruff, E.F., et. al. (2015) Biomolecules 5(2):1035-1062.
In bacterial systems, the sigma factor locates the transcriptional start site using key DNA sequence elements located at -35 nucleotides and -10 nucleotides from the transcriptional initiation site (Fig 10.7A) For RNAP from Thermus aquaticus, the −35 element interacts exclusively with σA4. The duplex DNA just upstream of the −10 element (−17 to −13) interacts with β′, σA3, and σA2 (Fig. 10.7B). Flipping of the A−11(nt) base from the duplex DNA into its recognition pocket in σA2 is thought to be the key event in the initiation of promoter melting and the formation of the transcription bubble (Figure 107.C). Once the transcription bubble has formed and transcription initiates, the sigma subunits dissociate from the complex and the RNAP catalytic subunit continues elongation on its own.
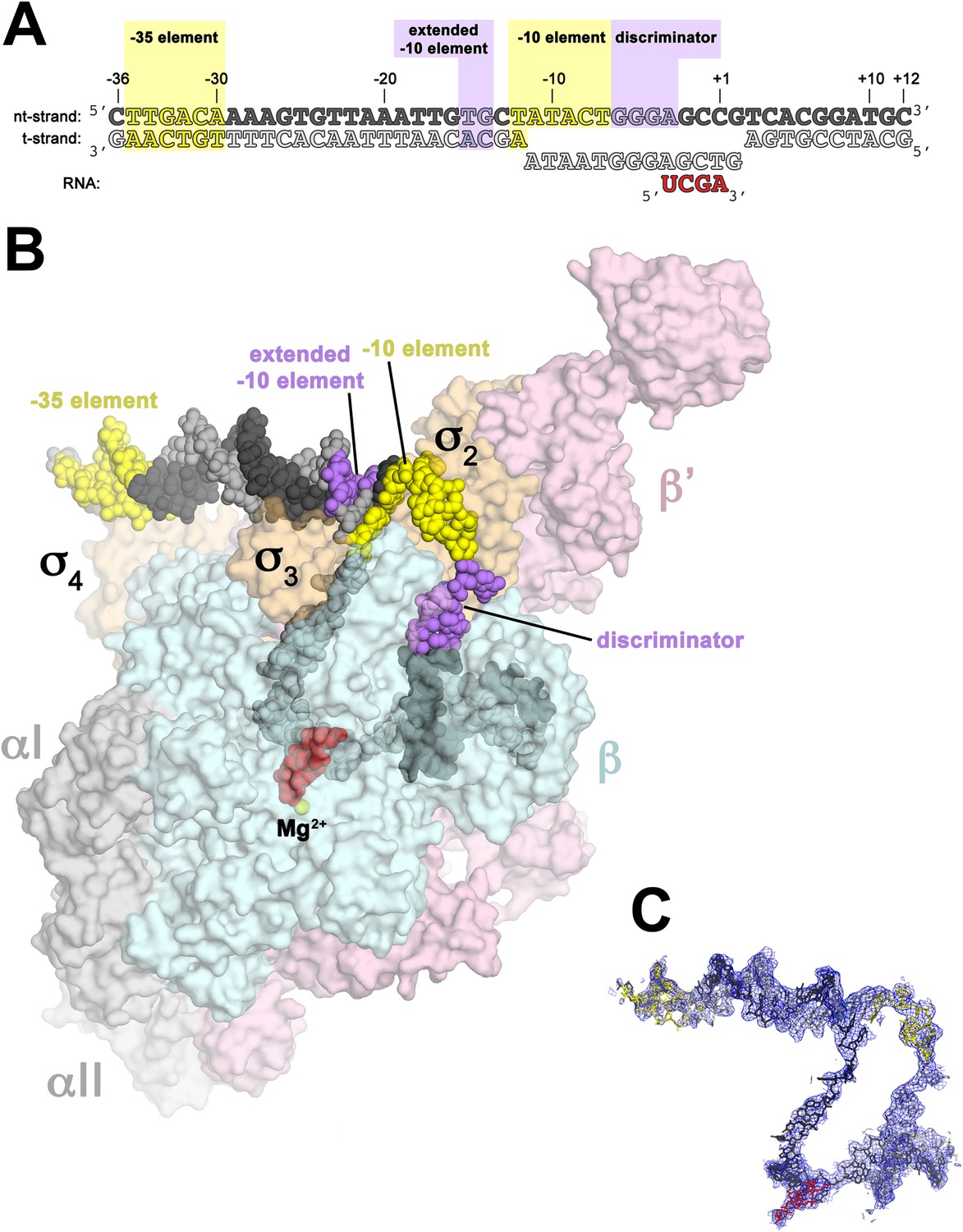
Figure 10.7 Structure of the RNAP Holoenzyme in Thermus aquaticus. (A) Oligonucleotides used for the crystallization of the RNAP holoenzyme in the open conformation. The numbers above denote the DNA position with respect to the transcription start site (+1). The −35 and −10 (Pribnow box) elements are shaded yellow, the extended −10 and discriminator elements purple. The nontemplate-strand DNA (top strand) is colored dark grey; template-strand DNA (bottom strand), light grey; RNA transcript, red. (B) Overall structure of RNAP holoenzyme in the open conformation bound with the DNA nucleotides. The nucleic acids are shown as CPK spheres and color-coded as in diagram A. Within RNAP, the αI, αII, ω, are shown in grey; β in light cyan; β′ in light pink; Δ1.1σA in light orange. The Taq EΔ1.1σA is shown as a molecular surface and the forward portion of the RNAP holoenzyme is transparent to reveal the RNAP active site Mg2+ (yellow sphere) and the nucleic acids held inside the RNAP active site channel. (C) Electron density and model for RNAP holoeznzyme nucleic acids in the open conformation. Color coding matches diagram A.
Eukaryotic RNA Polymerase Enzymes
In eukaryotic cells, three RNAPs share the task of transcription, the first step in gene expression. RNA Polymerase I (Pol I) is responsible for the synthesis of the majority of rRNA transcripts, whereas RNA Polymerase III (Pol III) produces short, structured RNAs such as tRNAs and 5S rRNA. RNA Polymerase II (Pol II) produces all mRNAs and most regulatory and untranslated RNAs.
The three eukaryotic RNA polymerases contain homologs to the the five core subunits found in prokaryotic RNAPs. In addition, the eukaryotic Pol I, Pol II and Pol III have five additional subunits forming a catalytic core that contains 10-subunits (Fig. 10.8). The core has a characteristic crab-claw shape which encloses a central cleft that harbors the DNA, and has two channels, one for the substrate NTPs and the other for the RNA product. Two ‘pinchers’, called the ‘clamp’ and ‘jaw’ stabilize the DNA at the downstream end and allow opening and closing of the cleft. For transcription to occur, the enzyme has to maintain a transcription bubble with separated DNA strands, facilitate the addition of nucleotides, translocate along the template, stabilize the DNA:RNA hybrid and finally allow the DNA strands to reanneal. This is achieved by a number of conserved elements in the active site, which include the fork loop(s), rudder, wall, trigger loop and bridge helix.
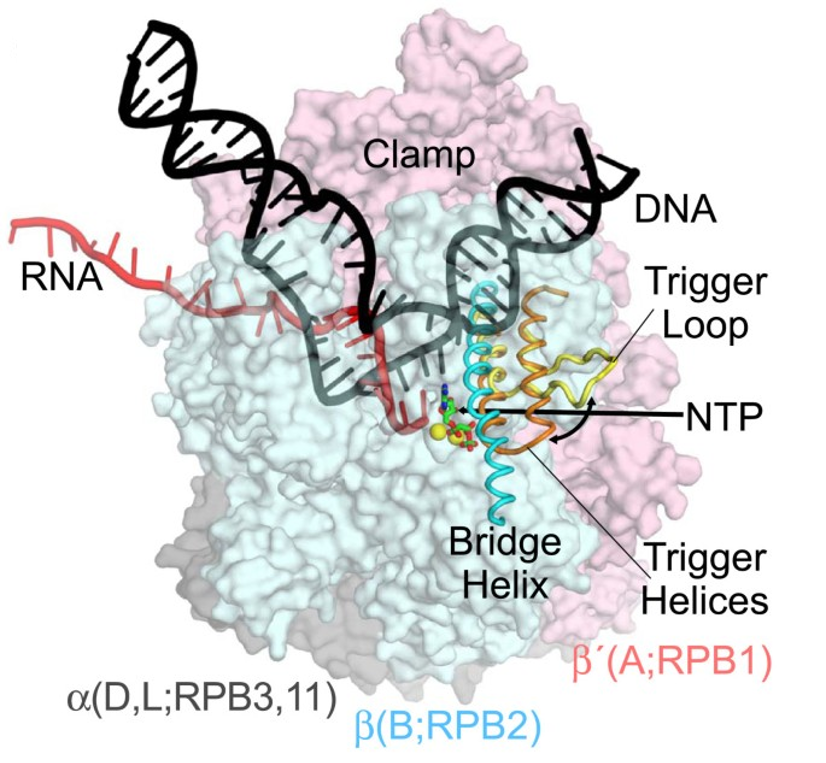
Figure 10.8 Structural Diagram of RNA Polymerase from Thermus thermophilus (PDB205j). DNA (black) is melting into a transcription bubble that allows template-strand pairing with RNA (red) in a 9-10 base pair RNA-DNA hybrid. The bridge helix (cyan) and trigger loop/helices (yellow/orange) lie on the downstream side of the active site. The presumed path of NTP entry is indicated by the straight arrow. Interconversion of the trigger loop and trigger helices is indicated by the curved arrow. The RNA polymerase subunits are shown as semi-transparent surfaces with the identities of orthologous subunits in bacteria (α, β, and β’, gray, blue, and pink, respectively), archaea (D, L, B, and A), and eukaryotic RNA polymerase II (RPB3, 11, RPB2, RPB1) indicated. The active site Mg2+ ions are shown as yellow spheres, and α,β-methylene-ATP in green and red.
Figure from: Hein, P.P. and Landick, R. (2010) BMC Biology 8:141
Apart from the core elements, all eukaryotic RNAPs share two additional, more distantly related subunits that form the stalk. In Pol I and Pol III, the core is further decorated with peripheral subunits grouped as heterodimeric (Pol Iand Pol III) and heterotrimeric (Pol III) subcomplexes. Accordingly, Pol I and Pol III holoenzymes contain 14 and 17 subunits, respectively, compared to the 12-subunit Pol II enzyme (Fig 10.9). The peripheral subunits of Pol I and Pol III have been suggested to be homologous to general Pol II transcription factors based on sequence similarity and the location of these subunits on the RNAP core. The Pol Iand Pol III heterodimers are homologous to TFIIF and likewise, the Pol III heterotrimer to TFIIE leading to the notion that during evolution, Pol I and Pol III stably integrated these transcription factor-like subunits into the core enzyme. Pol II, on the otherhand, associates more transiently with general transcription factors that form a preinitiation complex (PIC) and shown in Figure 10.9.

Figure 10.9 Comparison of the structures of Pol I, Pol II and Pol III demonstrating similar positions of Pol I- and Pol III-specific subunits compared to TFIIF and TFIIE in the Pol II-PIC. All structures were superimposed onto the largest subunit and depicted in the same orientation. Corresponding subunits are shown in the same color. Pol II-PIC components without counterparts in the Pol I and Pol III structures are depicted in grey. PDB IDs 4c3j, 5c4x, 5fj8 and 5fyw.
Figure from: Khatter, H., Vorlander, M.K., and Muller, C.W. (2017) Current Opinions in Structural Biology 47:88-94.
Back to the Top
10.3 Transcription Factors and the Preinitiation Complex (PIC)
Unlike prokaryotic systems which can initiate the recruitment of RNAP holoenzymes directly onto the DNA promoter regions and mediate the conversion of RNAP to the open conformation, eukaryotic RNA polymerases require a host of additional general transcription factors (GTFs), to enable this process. Here we will focus on the activation of RNA Polymerase II as an example of the complexity of eukaryotic transcription initiation.
Class II gene transcription in eukaryotes is a tightly regulated, essential process controlled by a highly complex multicomponent machinery. A plethora of proteins, more than a hundred in humans, are organized in often very large multiprotein assemblies that include a core of General Transcription Factors (GTFs). The GTFs include the factors TFIIA, TFIIB, TFIID, TFIIE, TFIIF, TFIIH, RNA polymerase (RNA pol II), as well as a large number of diverse complexes that act as co-activators, co-repressors, chromatin modifiers and remodelers (Fig. 10.10). Class II gene transcription is regulated at various levels: while assembling on chromatin, before and during transcription initiation, throughout elongation and mRNA processing, and termination. A host of activators and repressors has been reported to regulate transcription, including a central multisubunit complex called the Mediator that helps in the recruitment of GTFs and the activation of RNA Pol II. Here we will focus on the formation of the GTFs that make up the core preinitiation complex (PIC) during transcriptional activation.

Figure 10.10 Transcription Preinitiation Complex (PIC). Class II gene transcription in humans is brought about by over a hundred polypeptides assembling on the core promoter of protein-encoding genes, which then give rise to a mRNA. A PIC on a core promoter is shown in a schematic representation. PIC contains, in addition to promoter DNA, the GTFs (TFIIA, B, D, E, F, and H), and RNA Pol II. PIC assembly is thought to occur in a highly regulated, stepwise fashion, as indicated. TFIID is among the first GTFs to bind the core promoter via its TATA-box Binding Protein (TBP) subunit. Nucleosomes at transcription start sites contribute to PIC assembly, mediated by signaling through epigenetic marks on histone tails. The Mediator (not shown) is a further central multiprotein complex identified as a global transcriptional regulator. TATA = TATA-box DNA; BREu = B recognition element upstream; BREd = B recognition element downstream; Inr = Initiator; DPE = Down-stream promoter element.
Figure from: Gupta, K., et.al. (2016) J Mol Bio 428(12):2581-91.
Transcription of RNA pol II-dependent genes is triggered by the regulated assembly of the Preinitiation Complex (PIC). PIC formation commences with the binding of TFIID to the core promoter. TFIID is a large megadalton-sized multiprotein complex with around 20 subunits made up of 14 different polypeptides: the TATA-box binding protein (TBP) and the TBP-associated factors (TAFs) (numbered 1–13) (Fig. 10.11). Some of the TAF subunits are present in two copies. A key feature in TAFs is the histone fold domain (HFD), which is present in 9 out of 13 TAFs in TFIID. The HFD is a strong protein–protein interaction motif that mediates specific dimerization (Fig 10.11). The HFD-containing TAFs are organized in discrete heterodimers, with the exception of TAF10, which is capable of forming dimers with two different TFIID components, TAF3 and TAF8. HFDs and several other structural features of TBP and the TAFs are well conserved between species.

Figure 10.11 Human TFIID. TFIID is a large megadalton-sized multiprotein complex comprising about 20 subunits made up of 14 different polypeptides. The constituent proteins of TFIID, TBP and the TAFs, are shown in a schematic representation depicted as bars (inset, left). Structured domains are marked and annotated. The presumed stoichiometry of TAFs and TBP in the TFIID holo-complex is given (far left, gray underlaid). TAF10 (in italics) makes histone fold pair separately with both TAF3 and TAF8. TAFs present in a physiological TFIID core complex extracted from eukaryotic nuclei are labeled in bold. The architecture of TFIID core complex (EMD-2230) determined by cryo-EM is shown (bottom left) in two views related by a 90° rotation (arrows) [35]. The holo–TFIID complex is characterized by remarkable structural plasticity. Two conformations, based on cryo-EM data (EMD-2284 and EMD-2287), are shown on the right, a canonical form (top) and a more recently observed rearranged form (bottom). In the rearranged conformation, lobe A (colored in red) migrates from one extreme end of the TFIID complex (attached to lobe C) all the way to the other extremity (attached to lobe B)
Figure from: Gupta, K., et.al. (2016) J Mol Bio 428(12):2581-91.
TFIID was shown to adopt an asymmetric, horse-shoe shape with three almost equal-sized lobes (A, B, and C), exhibiting a considerable degree of conformational flexibility with at least two distinct conformations (open and closed) (Fig.10.11). The TBP component of TFIID binds with a specific DNA sequenced called the TATA box. This DNA sequence is found about 30 base pairs upstream of the transcription start site in many eukaryotic gene promoters. When TBP binds to a TATA box within the DNA, it distorts the DNA by inserting amino acid side-chains between base pairs, partially unwinding the helix, and doubly kinking it. The distortion is accomplished through a great amount of surface contact between the protein and DNA. TBP binds with the negatively charged phosphates in the DNA backbone through positively charged lysine and arginine amino acid residues. The sharp bend in the DNA is produced through projection of four bulky phenylalanine residues into the minor groove. As the DNA bends, its contact with TBP increases, thus enhancing the DNA-protein interaction. The strain imposed on the DNA through this interaction initiates melting, or separation, of the strands. Because this region of DNA is rich in adenine and thymine residues, which base-pair through only two hydrogen bonds, the DNA strands are more easily separated.
TFIID binding to the core promoter TATA box involves the release of TBP from Lobe A, likely in stages that overcome each of the inhibitory interactions blocking different functional surfaces of TBP (Figure 10.12). The interactions of the TAND1 and TAND2 regions of TAF1 with TBP (Figure 10.12a,d) are likely to be released (i) as Lobe C engages downstream DNA and positions the upstream promoter region such that it can be ‘scanned’ by TBP (Figure 10.12b,e), and (ii) as TFIIA joins the complex via interaction with Lobe B and further stabilizes the location of TBP for DNA interaction (Figure 10.12c,f). The final engagement of TBP with the promoter, with the concomitant bending of the DNA, is sterically incompatible with the TBP-TAF11 interaction, and therefore leads to TBP detachment from Lobe A and the opening of the binding site for TFIIB (Figure 10.12f), which in turn will engage Pol II, likely bound to TFIIF, thus promoting PIC assembly (Figure 10.12g).

Figure 10.12 Changes in TBP binding partners through the process of promoter binding. On the top are the atomic structures of human TFIID in the process of promoter engagement. With (a) in the canonical state, (b) in the scanning state and (c) in the engaged state. On the bottom, (d) shows TBP bound by inhibitory TAFs, (e) how these TAFs act to inhibit DNA binding and (f) how TBP binds the general transcription factors TFIIA and TFIIB when in complex with the PIC. The engaged TFIID complex recruits Pol II (g) with the aid of TFIIB and TFIIF.
Figure from: Patel, A.B., et. al. (2020) Curr Op Struc Bio 61:17-24.
Thus, the binding of TFIID to the core-promoter is followed by the recruitment of further GTFs and RNA pol II. Several lines of evidence suggest that this process occurs in a defined, stepwise order and undergoes significant restructuring. First, PIC adopts an inactive state, the “closed” complex, which is incompetent to initiate transcription. In addition to TFIID, TFIIH is also critical for the shift of RNA Pol II from the closed to the open conformation. TFIIH has an ATP-dependent translocase activity within one of its subunits, that opens up about 11 to 15 base pairs around the transcription start site by moving along one DNA strand inducing torsional strain, leading to conformational rearrangements and the positioning of single-stranded DNA to the active site of RNA pol II. In this “open” complex, RNA pol II can enter elongation to transcribe throughout a gene in a highly processive manner without dissociating from the DNA template or losing the nascent RNA.
In most eukaryotes, after synthesizing about 20–100 bases, RNA pol II can pause (Promoter proximal pause) and then disconnect from promoter elements and other components of the transcription machinery, giving rise to a fully functional elongation complex in a process called promoter escape. The promoter-bound components of the PIC, in contrast, remain in place, and thus only TFIIB, TFIIF, and RNA pol II need to be recruited for re-initiation, significantly increasing the transcription rate in subsequent rounds of transcription. Promoter escape is preceded by an abortive transcription in many systems, where multiple short RNA products of 3 to 10 bases in length are synthesized.
In addition to promoter elements within the DNA, enhancer elements are also important for the initiation of transcription. Promoters are defined as DNA elements that recruit transcription complexes for the synthesis of coding and non-coding RNA. Enhancers are defined as DNA elements that positively regulate transcription at promoters over long distances in a position- and orientation-independent manner. However, studies have revealed that many enhancers can recruit Pol II and initiate transcription of enhancer RNA (eRNA), thus blurring the functional distinction between enhancers and promoters (Figure 10.13).
Enhancer transcription produces relatively short ncRNA. Furthermore, transcription at enhancers is unstable and often leads to abortion of elongation. In contrast, transcription initiation at most Pol II promoters is stable and produces long mRNAs. Topological studies revealed that enhancers come in close proximity to target gene promoters during transcription activation. According to current gene activation models, the Mediator complex forms a physical bridge between distant regulatory regions and promoters, thereby promoting looping. Transcription of at least a subset of genes regulated by enhancers occurs in bursts indicating a discontinuous process of transcription complex recruitment, assembly, and/or conversion to elongation competent forms. The bursting phenomenon suggests that enhancer/promoter contacts may be transient and infrequent (Fig 10.13).
![]()
Figure 10.13 RNA polymerase II Enhancer Transfer Model. Depicted are steps involved in the recruitment of Pol II to SEs, assembly into elongation competent transcription complexes, transcription initiation and elongation, abortion and termination, and transfer to target genes. Transcription factors recruit Mediator and other co-regulators to SEs. Mediator recruits Pol II and assembles a fraction into elongation competent transcription complexes. Transcription is initiated by phosphorylation of the CTD. Early abortion and transcription termination conferred by Integrator releases Pol II, which is dephosphorylated and transferred to target gene promoters. Super Enhancer Element (SE);
Figure from: Gurumurthy, A., et.al. (2018) BioEssays 1800164
Back to the Top
10.4 Transcriptional Elongation and Termination
Prokaryotic Transcriptional Elongation
The rate of transcription elongation by E. coli RNAP is not uniform. RNA synthesis is characterized by pauses, some of which may be brief and resolved spontaneously, whereas others may lead to the transcription elongation complex (TEC) backtracking.
Elongation rate and pausing are determined by template sequence and RNA structure (e.g., stem-loops) and involve at least two components of the RNAP catalytic center, the bridge helix (BH) and trigger loop (TL). Elongation is proposed to occur in three steps (Fig. 10.14). First, the TL folds in response to NTP binding. Mutational analyses indicate that this conformational change in the TL can be rate-limiting, and reflects the ability of the incoming NTP to bind to TEC. The second step is the incorporation of the NTP and the release of pyrophosphate. The third step involves the translocation of the RNAP down the DNA Template such that the next RNA nucleotide can be added to the nascent transcript.

Figure 10.14 A model for Transcriptional Elongation. The trigger loop hinges, bridge helix hinges and bridge helix bending models are based on all atom molecular dynamics simulations. At the top of the figure, diagrams of the closed TEC, the closed product TEC (after chemistry) and the translocating TEC are shown. DNA is grey; RNA is red; the NTP substrate (or incorporated NMP and pyrophosphate) is blue; the trigger loop (TL) is purple; the bridge helix (BH) is yellow. Interpretations of simulations are shown schematically below. Simulations indicate trigger loop hinges H1 and H2, bridge helix hinges H3 and H4 and bridge helix bend modes B1 (straighter) and B2 (more sharply bent).
Figure from: Kireeva, M., et. al. (2012) BMC Biophysics 5(1):11
Backtracking of TEC may take place after a brief pause in transcription, caused by the thermodynamic properties of nucleic acids sequences surrounding the elongation complex. In addition, misincorporation events render elongation complexes prone to backtracking by at least one bp. In this case, the rescue from backtracking through the cleavage of the 3′ end of the erroneous transcript also may be seen as a proofreading reaction. Any backtracking event causes a pause or arrest of transcription elongation, which may limit its overall rate (the average speed of RNAP along the template) or processivity (the fraction of RNAP molecules reaching the end of the gene).
While the general structure of the elongation complex (the transcription bubble, the RNA-DNA hybrid) remains unchanged during backtracking, extension of RNA becomes impossible in this conformation. However, such complexes can be resolved by the hydrolytic activity of RNAP, which cleaves the phosphodiester bond in the active center of the backtracked complex, producing a new RNA 3′ end in the active center. For single base backups, the hydrolytic reaction is catalyzed by a flexible domain of RNAP located in the secondary channel called the Trigger Loop (TL; Figure 10.15B) and the two metal ions of the active center.
Longer sequences of backtracked TEC can restart when acted upon by GreA/B factors, which restore the 3′-end of the nascent transcript to the active center. GreA and GreB are transcript cleavage factors that act on backtracked elongation complexes. When Gre factors are bound in the secondary channel, Gre factors displace the TL from the active center (Figure 10.15). The displacement switches off the relatively slow TL-dependent intrinsic transcript hydrolysis, and imposes the highly efficient Gre-assisted hydrolysis. This efficiency is thought to be due to stabilization of the second catalytic Mg2+ ion and an attacking water molecule by the Gre factors.
![]()
Figure 10.15 The Role of Gre Factors in Relieving Transcription Elongation Complex Backtracking. (A) Ribbon diagram of the GreA and GreB proteins. (B) The mode of functioning of Gre factors. The Gre factor is bound to the active elongation complex but does not impose hydrolytic activity on it. Upon backtracking or misincorporation, the Gre factor protrudes its coiled-coil domain through the secondary channel of RNAP (shown in the lefthand diagram), where it substitutes for the catalytic domain Trigger Loop (TL). This substitution switches off the slow TL-dependent phosphodiester bond hydrolysis and, and instead, facilitates highly efficient Gre-dependent hydrolysis. After resolution of the backtracked complex through RNA cleavage, the elongation complex returns to the active conformation and the Gre factor gives way to the TL, which can now continue catalysis of RNA synthesis (shown in the righthand diagram). The controlled switching between Gre and the TL eliminates possible interference of Gre with the RNA synthesis.
Figure modified from: Zenkin, N., and Yuzenkova, Y. (2015) Biomolecules 5(3):1195-1209.
Back to the Top
Prokaryotic Transcriptional Termination
Transcription termination determines the ends of transcriptional units by disassembling the transcription elongation complex (TEC), thereby releasing RNA polymerases and nascent transcripts from DNA templates. Failure in termination causes transcription readthrough, which yields wasteful and possibly harmful intergenic transcripts. It can also perturb expression of downstream genes when the unterminated TEC sweeps transcription initiation complexes off their promoters or collides with RNA polymerases that transcribe opposite strands.
Transcriptional termination in prokaryotes can be template-encoded and factor-independent (intrinsic termination), or require accessory factors, such as Rho, Mfd and DksA. Intrinsic termination occurs at specific template sequences – an inverted repeat followed by a run of A residues. Termination is driven by formation of a short stem-loop structure in the nascent RNA chain (Figure 10.16). RNA synthesis arrests and TEC dissociates at the 7th and 8th U of the run. Formation of the stem-loop dissociates the weak rU:dA hybrid. Stem-loop formation is hindered by upstream complementary RNA sequences that compete with the downstream portion of the stem, as well as by RNA: protein interactions in the RNA exit channel. Intrinsic termination depends critically upon timing. Hairpin folding and transcription of the termination point must be coordinated, so that the complete hairpin is formed by the time RNAP transcribes the termination point. The size of the stem, the sequence of the stem and the length of the loop all affect termination efficiency.
The bridge α-helix in the β’ subunit borders the active site and may have roles in both catalysis and translocation. Mutations in the YFI motif (β’ 772-YFI-774) affect intrinsic termination as well as pausing, fidelity and translocation of RNAP. One mutation, F773V, abolishes the activity of the λ tR2 intrinsic terminator, although neighboring mutations have little affect on termination. Modeling suggests that this unique phenotype reflects the ability of F773 to interact with the fork domain in the β subunit.

Figure 10.16 Model of Intrinsic Termination. (A) Shows the open conformation of the RNAP during transcriptional elongation. RNAP is shown in yellow, DNA template in blue, and nascent RNA in red. Key elements of the RNAP RNA exit channel are shown in grey and labeled as indicated. (B) Shows the extension of the nascent RNA through the RNAP exit channel and the potential for forming the RNA hairpin structure when enough length has been achieved. (C) Shows the clamp opening and disintegration of the the TEC when the RNA hairpin structure is encountered at the transcriptional bubble.
Figure from: Washburn, R.S., and Gottesman, M.E. (2015) Biomolecules 5(2)1063-1078.
Transcriptional termination can also be dependent upon accessory factors, such as the Rho protein. Transcription termination factor Rho is an essential protein in E. coli first identified for its role in transcription termination at Rho-dependent terminators, and is estimated to terminate ~20% of E. coli transcripts. The rho gene is highly conserved and nearly ubiquitous in bacteria. Rho is an RNA-dependent ATPase with RNA:DNA helicase activity, and consists of a hexamer of six identical monomers arranged in an open circle (Figure 10.17A).
Rho binds to single stranded RNA in a complex multi-step pathway that involves two distinct sites on the hexamer. The primary binding site (PBS), distributed on the N-terminal domains around the hexamer (Figure10.17B, cyan), ensures initial anchoring of Rho to the transcript at a Rut (Rho utilization) site, a∼70 nucleotides (nt) long, cytidine-rich and poorly-structured RNA sequence. Each Rho monomer contains a subsite capable of binding specifically the base residues of a 5′-YC dimer (Y being a pyrimidine). Biochemical and structural data suggest that Rho initially binds to RNA in an open, ‘lock-washer’ conformation that closes into a planar ring as RNA transfers to the central cavity. There, the ssRNA contacts an asymmetric secondary binding site (SBS) (Fig. 10.17B, green), and this step, which presumably is rate-limiting for the overall reaction, leads to motor activation. Upon hydrolysis of ATP, the ssRNA is pulled upon conformational changes of the conserved Q and R loops of the SBS, leading to Rho translocation, and ultimately promoting RNA polymerase (RNAP) dissociation. The molecular mechanism of Rho translocation based on single-molecule fluorescence methods appears to be tethered tracking. The tethered tracking model postulates that Rho maintains its contacts between the PBS and the loading (Rut) site upon translocation (Figure 10.17B). This mechanism would allow Rho to maintain its high affinity interaction with Rut, and implies the growing of an RNA loop between the PBS and the SBS upon translocation (Fig 10.17B).
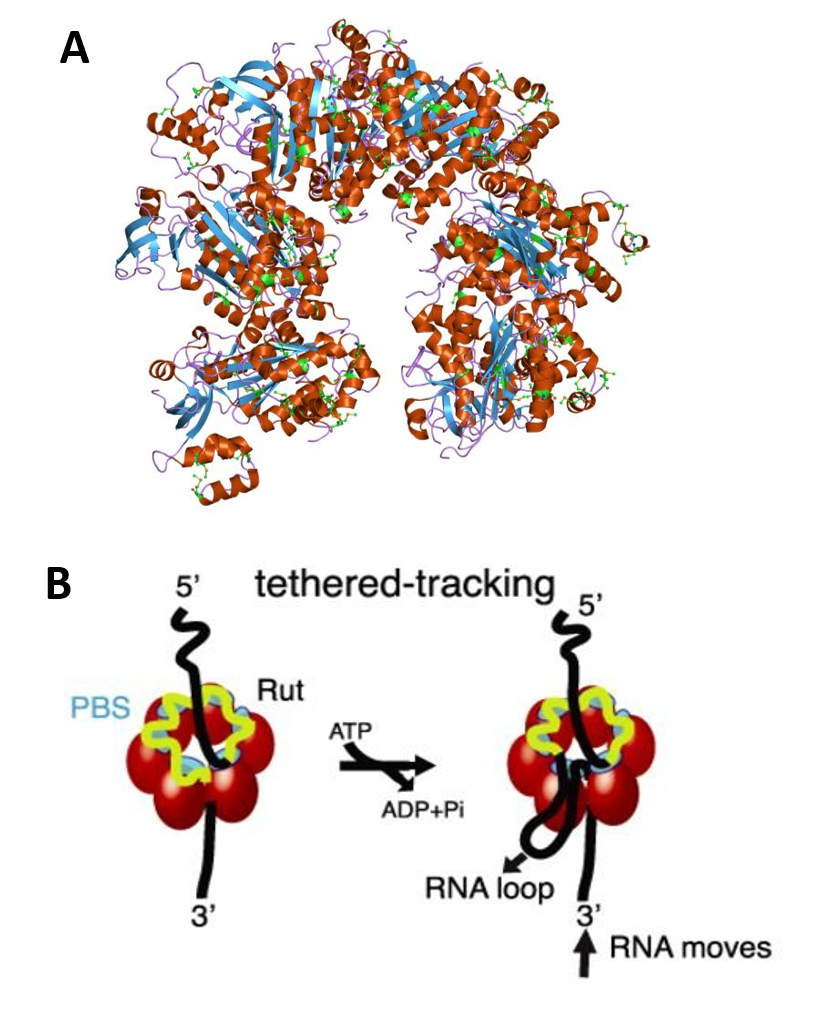
Figure 10.17 Schematic of Rho factor structure and mechanisms. (A) Molecular structure of the Rho protein (PDB 1pv4) (B) Rho assembles as a homo-hexameric ring (red spheres or tetragons), with RNA (black/yellow curve) binding to the primary binding sites (PBS, cyan) and the secondary binding sites inside the ring (SBS, green), where ATP-coupled translocation takes place. The Rut specific binding site is depicted in yellow. The tethered-tracking model proposed that Rho translocates RNA while maintaining interactions between PBS and Rut. This model requires the formation of a loop that would shorten the extension of RNA upon translocation.
Figure modified from: (A) Jawahar Swaminathan and (B) Gocheva, V., et. al. (2015) Nuc Acids Res 43(1)10.1093.
{kind=link}
Back to the Top
Eukaryotic Transcriptional Termination
In eukaryotes, termination of protein-coding gene transcription by RNA polymerase II (Pol II) usually requires a functional polyadenylation (pA) signal, typically a variation of the AAUAAA hexamer. Nascent pre-mRNA is cleaved and the 5′ fragment is polyadenylated at the pA site shortly downstream from the hexamer by cleavage and pA factors (CPFs). Two mechanisms have been suggested for pA-dependent transcription termination. In the allosteric model, the pA signal and/or other termination signals bind with the pA signal downstream region (PDR) and induce reorganization of the Pol II complex. This includes the association or dissociation of endonuclease components such as the CPFs. This causes conformational changes in Pol II and TEC disassembly ensues. In the kinetic model, also known as the “torpedo” model, cleavage at the pA site separates the pre-mRNA from the TEC, which continues synthesizing a downstream nascent transcript. This new transcript is a substrate of XRN2/Rat1p, a processive 5′-to-3′ exoribonuclease that catches up with, and disassembles, the TEC by an unknown mechanism.
The two pA-dependent models are not mutually exclusive, and unified models have been proposed. Loosely conserved pA signal sequences downstream of protein-coding genes bind to components of the polyadenylation factor (CF1) complex leading to assembly of the cleavage and polyadenylation machinery. Termination is coupled to cleavage in a manner that has not yet been completely resolved, however, one of the major factors involved in yeast pA termination is the endonuclease, Ysh1. For example, the depletion of Ysh1 blocks TEC dissociation, but does not cause substantial readthrough at the termination site (Fig. 10.18 A&B). These results suggest that Ysh1 does not directly cause the pausing that occurs in the allosteric termination pathway, but rather plays a role in the dissociation of the Pol II complex from the DNA template (Figure 10.18A & B). It should be noted that not all pA-dependent termination is dependent on Ysh1 and that other mechanisms of pA-mediated termination still remain to be elucidated.

Figure 10.18 Schematic representation of Pol II termination after removal of non-pA and pA termination factors. Elongating Pol II (green) terminates pA transcripts (A) after an allosteric change (red) that reduces processivity. (B) Depletion of Ysh1 leads to minimally extended readthrough transcripts but does not block the allosteric change in Pol II. (C) Nrd1 and Nab3 binding recruit Sen1 for termination of non-pA transcripts. (D) Pol II elongation complex lacking Nrd1 does not recognize termination sequences in the nascent transcript and thus does not facilitate the allosteric transition in Pol II. This leads to processive readthrough. (E) Nrd1 and Nab3 recognize terminator sequences allowing the allosteric change in Pol II but depletion of Sen1 blocks removal of Pol II from the template.
Figure from: Schaughency, P., Merran, J., and Corden J.L. (2014) PLOS Genetics 10(10):e1004632
The mechanisms of termination of Pol II-mediated transcription differ for coding and non-coding transcripts. Coding transcripts and possibly some stable uncharacterized transcripts (SUTs) are nearly always processed at the 3′-end by the cleavage and polyadenylation (pA) machinery and are processed by the pA-dependent termination mechanisms described above. In contrast, ncRNAs are terminated and processed by an alternative pathway that, in yeast, requires the RNA-binding proteins Nrd1 and Nab3, as well as, the RNA helicase Sen1 (Fig 10.18 C). Nrd1 and Nab3 recognize RNA sequence elements downstream of snoRNAs and CUTs and this leads to the association of a complex that contains the DNA/RNA helicase Sen1 and the nuclear exosome. The nuclear exosome is a complex of ribonucleases with 3′ to 5′ exonuclease and endonuclease activity. It functions to degrade unstable or incorrect RNA transcripts.
Both Nrd1 and Sen1 depletion lead to readthrough transcription of ncRNAs, suggesting their importance in non-pA-dependent transcription termination (Fig 10.18 C & D). Furthermore, depletion of Nrd1 also causes the accumulation of longer readthrough ncRNAs, suggesting its role in trafficking ncRNAs to the nuclear exosome following termination.
Back to the Top
10.5 Processing of RNA
Post-transcriptional modifications of rRNA and tRNA will be topics of Chapter 11 as their structure and function in protein synthesis will be a focal point. Thus, this section will focus on post-transcriptional modifications of mRNA.
Prokaryotic RNA Processing
Bacterial cells do not have extensive post-transcriptional modification of mRNA primarily because transcription and translation are coupled processes. Bacterial cells lack the physical barrier of a nucleus, which allows transcription and translation machineries to function at the same time, enabling the concurrent translation of an mRNA while it is being transcribed (Fig 10.19). Within this system the NusG protein plays a critical role. NusG has three separate domains and the functions of two of them are known. The NusG N-terminal domain (NusG-NTD) has the capacity to bind to RNAP, whereas the C-terminal domain (NusG-CTD) can combine with the NusE (RpsJ) component of ribosomes. These two functions of NusG enable transcription to be coupled with translation. NusG CTD can also bind to Rho to terminate transcription (Figure 10.19).

Fig. 10.19. The roles of NusG in transcription/translation coupling. (a) Composition of an active RNAP complex. RNAP is shown in dark grey, DNA in blue and nascent RNA in red. The ribosome is shown in green with the nascent polypeptide chain in light grey; the bulge in the small subunit denotes the location of NusE (RpsJ). NusG is shown in orange: its shape denotes two functional sections. The larger section denotes the N-terminal domain, which binds to RNAP. The smaller section denotes the C-terminal domain, which interacts with NusE in situ. Rho is shown in purple. (b) After translation is completed NusG remains bound to RNAP and may also bind to Rho through the C-terminal domain leading to termination of transcription.
Figure from: Cortes, T., and Cox, R.A. (2015) Microbiology 161:719-728.
Eukaryotic RNA Processing
In multicellular organisms almost every cell contains the same genome, yet complex spatial and temporal diversity is observed in gene transcripts. This is achieved through multiple levels of processing leading from gene to protein, of which RNA processing is an essential stage. Following transcription of a gene by RNA polymerases to produce a primary mRNA transcript, further processing is required to produce a stable and functional mature RNA product. This involves various processing steps including RNA cleavage at specific sites, intron removal, called splicing, which substantially increase the transcript repertoire, and the addition of a 5’CAP. Another crucial feature of the RNA processing of most genes is the generation of 3′ ends through an initial endonucleolytic cleavage, followed in most cases by the addition of a poly(A) tail, a process termed 3′ end cleavage and polyadenylation (CPA).
3′-Polyadenylation
As seen in Section 10.4, polyadenylation is a required step for the correct termination of nearly all mRNA transcripts. With the exception of replication dependent histone genes, metazoan protein encoding mRNAs contain a uniform 3′ end consisting of a stretch of adenosines. In addition to deterimining the correct transcript length at transcription termination, the poly(A) tail helps to ensure the translocation of the nascent RNA molecule from the nucleus to the cytoplasm, enhances translation efficiency, acts as a signal feature for RNA degradation, and thereby contributes to the production efficiency of a protein.
CPA is carried out by a multi-subunit 3′ end processing complex, which involves over 80 trans-acting proteins, comprised of four core protein subcomplexes (Figure 10.20 A). These consist of (1) cleavage and polyadenylation specificity factor (CPSF), comprised of proteins CPSF1-4, factor interacting with PAPOLA and CPSF1 (FIP1L1), and WD repeat domain 33 (WDR33) (shown in green on Figure 10.20 A); (2) cleavage stimulation factor (CstF), a trimer of CSTF1-3 (shown in red on Figure 10.20 A; (3) cleavage factor I (CFI), a tetramer of two small nudix hydrolase 21 (NUDT21) subunits, and two large subunits of CPSF7 and/or CPSF6 (shown in orange in Figure 10.20 A); and (4) cleavage factor II (CFII), composed of cleavage factor polyribonucleotide kinase subunit 1 (CLP1) and PCF11 cleavage and polyadenylation factor subunit (PCF11) (shown in yellow on Figure 10.20 A). Additional factors include symplekin, the poly(A) polymerase (PAP), and the nuclear poly(A) binding proteins such as poly(A) binding protein nuclear 1 (PABPN1).
CPA is initiated by this complex recognising specific cis-element sequences within the nascent pre-mRNA transcripts termed polyadenylation signals (PAS). The PAS sequence normally consists of either a canonical AATAAA hexamer, or a close variant usually differing by a single nucleotide (e.g., ATTAAA, TATAAA). It is located 10 to 35 nucleotides upstream of the cleavage site (CS) usually consisting a CA dinucleotide. The PAS is also determined by surrounding auxiliary elements, such as upstream U-rich elements (USE), or downstream U-rich and GU-rich elements and G-rich sequences (DSE).
As soon as the nascent RNA molecule emerges from RNA polymerase II (RNA Pol II), the CPSF complex is recruited to the PAS hexamer through numerous interactions. Upon successful assembly of this macromolecular machinery, CPSF3 performs the endonucleolytic cleavage followed by a non-templated addition of approximately 50-100 A residues.
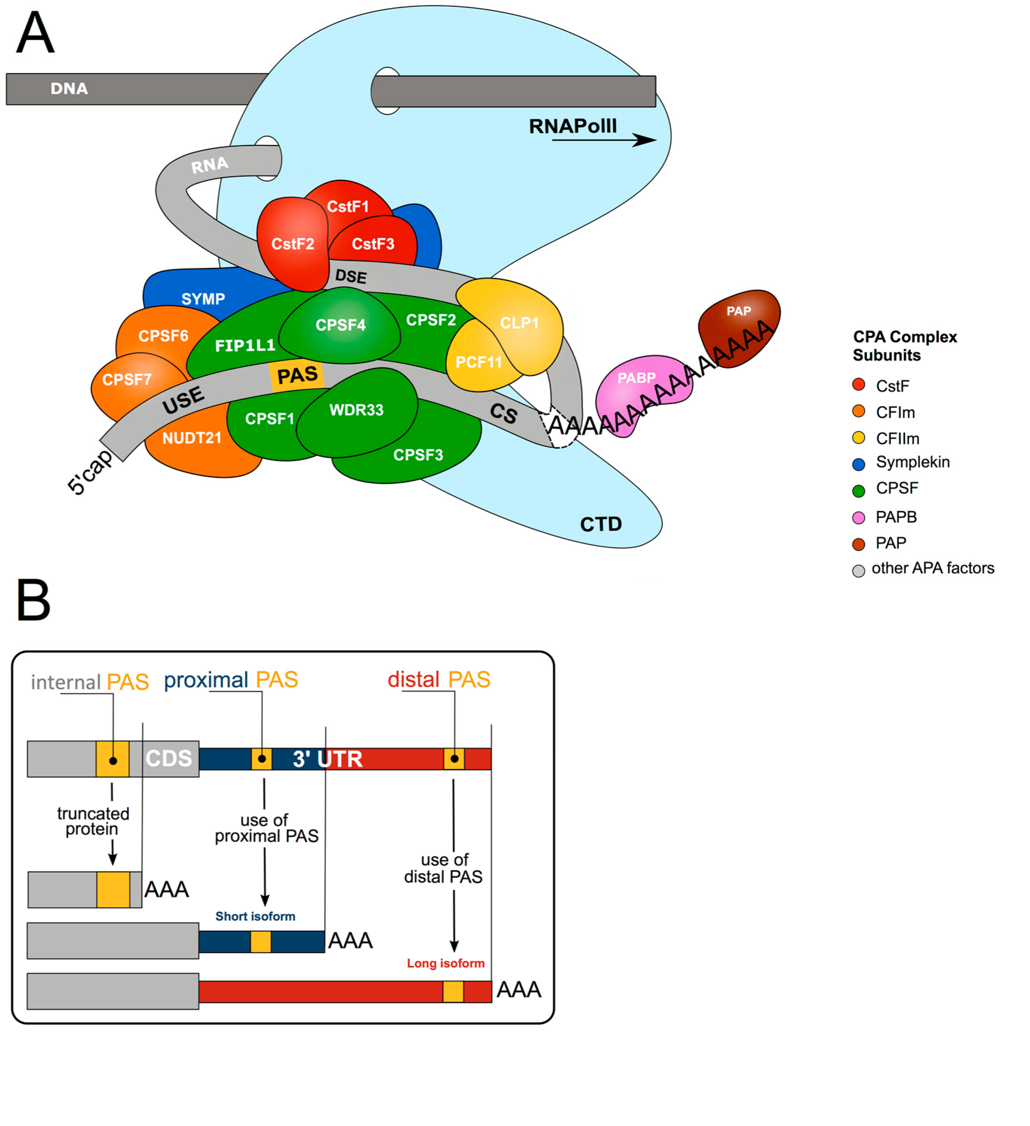
Figure 10.20. The core 3′ end RNA processing machinery and impact on alternative polyadenylation. (A) The core 3′ end processing machinery consists of complexes composed of multiple trans acting proteins interacting with RNA via multiple cis-elements (USE = upstream sequence element; PAS = poly(A) signal; CS = cleavage site; DSE = downstream sequence element; CTD = C-terminal domain). Upon co-transcriptional assembly of these complexes, RNA cleavage and polyadenylation occurs to form the 3′ end of the nascent RNA molecule. (B) More than 70% of all genes harbour more than one polyadenylation signal (PAS). This gives rise to transcript isoforms differing at the mRNA 3′ end. While alternative polyadenylation (APA) in 3′UTR changes the properties of the mRNA (stability, localisation, translation), internal PAS usage (in introns or the coding sequence (CDS)) changes the C-termini of the encoded protein, resulting in different functional or regulatory properties.
Figure from: Nourse, J., et. al. (2020) Biomolecules 10(6):915
Alternative polyadenylation (APA) occurs when more than one PAS is present within a pre-mRNA and provides an additional level of complexity in CPA-mediated RNA processing (Figure 10.20 B). Early studies revealed a significant portion of genes undergo APA, and with the advent of next-generation RNA sequencing technologies the large scale regulation of genes has become apparent, with approximately 70% of the transcriptome exhibiting APA regulation. As APA determines 3′UTR content and thus the regulatory features available to the mRNA, changes in the APA profile of a gene can have enormous impacts on expression.
5′-CAP Formation
In eukaryotes, the 5′ cap, found on the 5′ end of an mRNA molecule, consists of a guanine nucleotide connected to mRNA via an unusual 5′ to 5′ triphosphate linkage (Fig. 10.21). This guanosine is methylated on the 7 position directly after capping in vivo by a methyltransferase. It is referred to as a 7-methylguanylate cap, abbreviated m7G.
In multicellular eukaryotes and some viruses, further modifications exist, including the methylation of the 2′ hydroxy-groups of the first 2 ribose sugars of the 5′ end of the mRNA. Cap-1 has a methylated 2′-hydroxy group on the first ribose sugar, while cap-2 has methylated 2′-hydroxy groups on the first two ribose sugars. The 5′ cap is chemically similar to the 3′ end of an RNA molecule (the 5′ carbon of the cap ribose is bonded, and the 3′-OH unbonded). This provides significant resistance to 5′ exonucleases.
snRNAs contain unique 5′-caps. Sm-class snRNAs are found with 5′-trimethylguanosine caps, while Lsm-class snRNAs are found with 5′-monomethylphosphate caps. In bacteria, and potentially also in higher organisms, some RNAs are capped with NAD+, NADH, or 3′-dephospho-coenzyme A. In all organisms, mRNA molecules can be decapped in a process known as messenger RNA decapping.
For capping with 7-methylguanylate, the capping enzyme complex (CEC) binds to RNA polymerase II before transcription starts. As soon as the 5′ end of the new transcript emerges from RNA polymerase II, the CEC carries out the capping process (this kind of mechanism ensures capping, as with polyadenylation). The enzymes for capping can only bind to RNA polymerase II that is engaging in mRNA transcription, ensuring specificity of the m7G cap almost entirely to mRNA.

Figure 10.21 Structure of the 7-methylguanylate CAP.
Figure from: Brisbane
{kind=link}
The 5′ cap has four main functions:
- Regulation of nuclear export
- Prevention of degradation by exonucleases
- Promotion of translation (see ribosome and translation)
- Promotion of 5′ proximal intron excision
In addition to the polyA tail, nuclear export of RNA is regulated by the cap binding complex (CBC), which binds to 7-methylguanylate-capped RNA (Fig 10.22). The CBC is then recognized by the nuclear pore complex and the mRNA exported. Once in the cytoplasm after the pioneer round of translation, the CBC is replaced by the translation factors eIF4E and eIF4G of the eIF4F complex. This complex is then recognized by other translation initiation machinery including the ribosome, aiding in translation efficiency.
Capping with 7-methylguanylate prevents 5′ degradation in two ways. First, degradation of the mRNA by 5′ exonucleases is prevented by functionally looking like a 3′ end. Second, the CBC and eIF4E/eIF4G block the access of decapping enzymes to the cap. This increases the half-life of the mRNA, essential in eukaryotes as the export and translation processes take significant time.
The mechanism that promotes the 5′ proximal intron excision during splicing is not well understood, but the 7-methylguanylate cap appears to loop around and interact with the spliceosome, potentially playing a role in the splicing process.
Decapping of a 7-methylguanylate-capped mRNA is catalyzed by the decapping complex made up of at least Dcp1 and Dcp2, which must compete with eIF4E to bind the cap. Thus the 7-methylguanylate cap is a marker of an actively translating mRNA and is used by cells to regulate mRNA half-lives in response to new stimuli. During the decay process, mRNAs may be sent to P-bodies. P-bodies are granular foci within the cytoplasm that contain high levels of exonuclease activity.
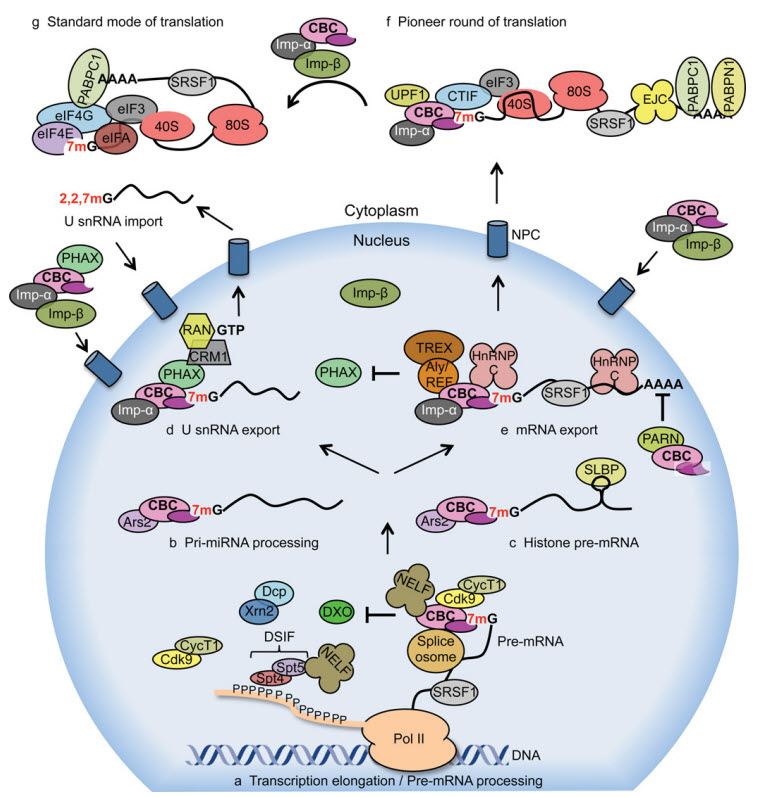
Figure 10.22 Importance of the 5-CAP during the lifespan of a mRNA Transcript (a) CBC is required for pre-mRNA processing. The co-transcriptional binding of CBC to 7mG prevents the decapping activities of pre-mRNA degradation complexes [DXO (decapping exoribonuclease) and Dcp (decapping mRNA) Xrn2 (5′–3′ exoribonuclease 2)] and promotes pre-mRNA processing. CBC recruits P-TEFb [Cdk9/Cyclin T1 (CycT1)] to transcription initiation sites of specific genes promoting phosphorylation of the RNA pol II CTD at Ser2 residues. This results in the recruitment of splicing factors including SRSF1, which regulates both constitutive and alternative splicing events. Furthermore, CBC interacts with splicing machinery components that results in the spliceosomal assembly. CBC interacts with NELF and promotes pre-mRNA processing of replication-dependent histone transcripts. (b) CBC forms a complex with Ars2 and promotes miRNA biogenesis by mediating pri-miRNA processing. (c) CBC/Ars2 promotes pre-mRNA processing of replication-dependent histone transcripts. (d) CBC promotes export of U snRNA. CBC interacts with PHAX, which recruits export factors including CRM1 and RAN·GTP. (e) CBC promotes export of mRNA. For export of transcripts over 300 nucleotides, hnRNP C interacts with CBC and inhibits the interaction between CBC and PHAX, allowing the CBC to interact with TREX and the transcript to be translocated to the cytoplasm. CBC interacts with the PARN deadenylase and inhibits its activity, protecting mRNAs from degradation. (f) CBC mediates the pioneer round of translation. Cbp80 interacts with CTIF, which recruits the 40S ribosomal subunit via eIF3 to the 5′ end of the mRNA for translation initiation. Upon binding of importin-β (Imp-β) to importin-α (Imp-α), mRNA is released from CBC and binds to eIF4E for the initiation of the standard mode of translation. CBC-bound mRNP components not found in eIF4E-bound mRNPs are CTIF, exon junction complex (EJC) and PABPN1. (g) The standard mode of translation is mediated by eIF4E cap-binding protein. eIF4E is a component of the eIF4F complex which promotes translation initiation.
Figure from: Gonatopoulos-Pournatzis, T. and Crowing V. (2014) Biochemical Journal 457(2):231-42.
Back to the Top
mRNA Splicing
Eukaryotic genes that encode polypeptides are composed of coding sequences called exons (ex-on signifies that they are expressed) and intervening sequences called introns (int-ron denotes their intervening role). Transcribed RNA sequences corresponding to introns do not encode regions of the functional polypeptide and are removed from the pre-mRNA during processing. It is essential that all of the intron-encoded RNA sequences are completely and precisely removed from a pre-mRNA before protein synthesis so that the exon-encoded RNA sequences are properly joined together to code for a functional polypeptide. If the process errs by even a single nucleotide, the sequences of the rejoined exons would be shifted, and the resulting polypeptide would be nonfunctional. The process of removing intron-encoded RNA sequences and reconnecting those encoded by exons is called RNA splicing. Intron-encoded RNA sequences are removed from the pre-RNA while it is still in the nucleus. Although they are not translated, introns appear to have various functions, including gene regulation and mRNA transport. On completion of these modifications, the mature transcript, the mRNA that encodes a polypeptide, is transported out of the nucleus, destined for the cytoplasm for translation. Introns can be spliced out differently, resulting in various exons being included or excluded from the final mRNA product. This process is known as alternative splicing. The advantage of alternative splicing is that different types of mRNA transcripts can be generated, all derived from the same DNA sequence. In recent years, it has been shown that some archaea also have the ability to splice their pre-mRNA.
The splicing reaction is catalyzed by the spliceosome, a macromolecular complex formed by five small nuclear ribonucleoproteins (snRNPs), termed U1, U2, U4, U5, and U6, and approximately 200 proteins (Fig. 10.23). The assembly of the spliceosome on pre-mRNA includes the binding of U1 snRNP, U2 snRNP, the pre-formed U4/U6-U5 triple snRNP, and the Prp19 complex. This assembly occurs through the recognition of several sequence elements on the pre-mRNA that define the exon/intron boundaries, which include the 5′ and 3′ splice sites (SS), the associated 3′ sequences for intron excision, the polypyrimidine (Py) tract, and the branch point sequence (BPS). The assembly of the spliceosome during the process is depicted in Figure 10.23.
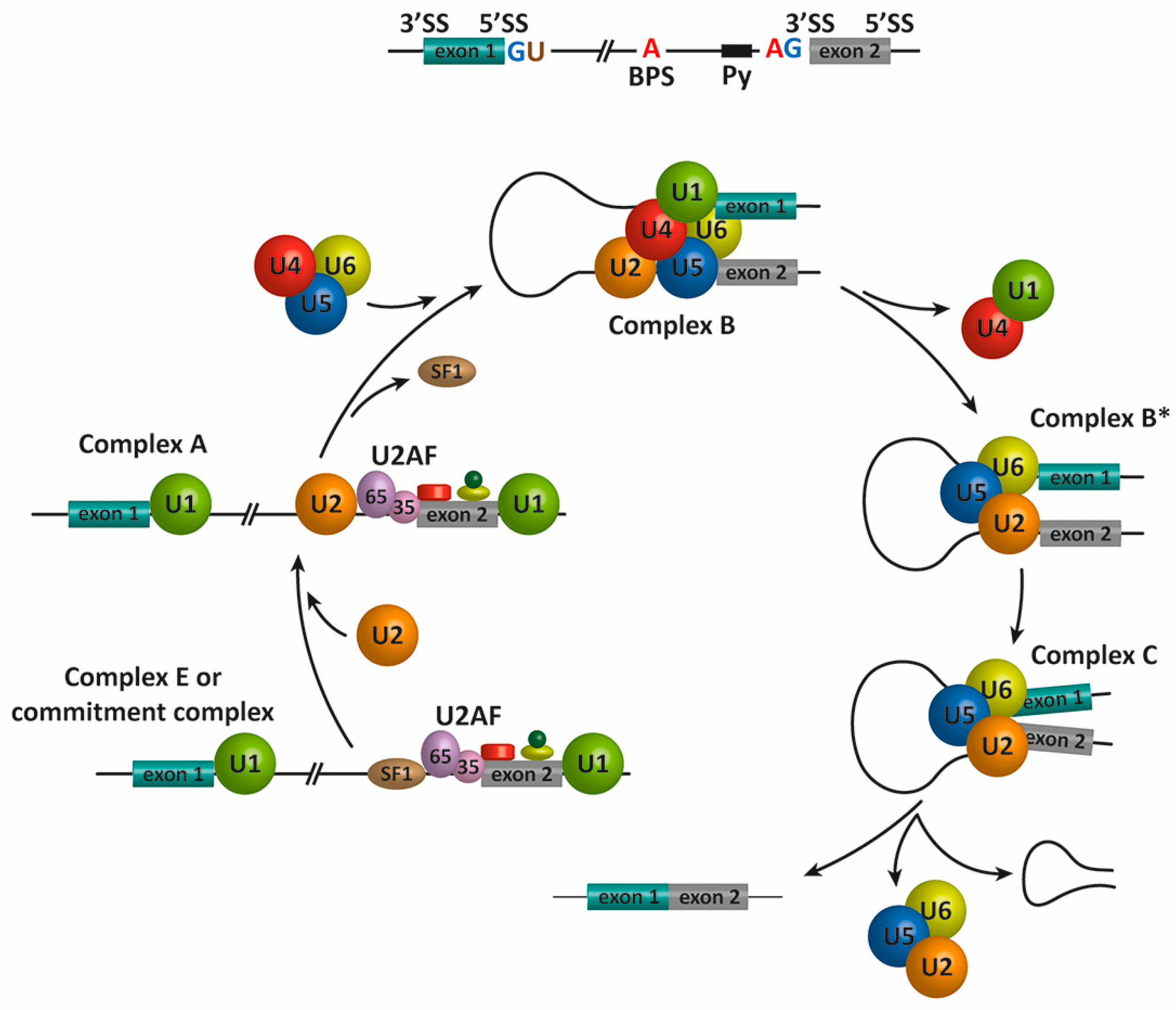
Figure 10.23 Schematic representation of the spliceosome assembly and pre-mRNA splicing. In the first step of the splicing process, the 5′ splice site (GU, 5′ SS) is bound by the U1 snRNP, and the splicing factors SF1/BBP and U2AF cooperatively recognize the branch point sequence (BPS), the polypyrimidine (Py) tract, and the 3′ splice site (AG, 3′ SS) to assemble complex E. The binding of the U2 snRNP to the BPS results in the pre-spliceosomal complex A. Subsequent steps lead to the binding of the U4/U5–U6 tri-snRNP and the formation of complex B. Complex C is assembled after rearrangements that detach the U1 and U4 snRNPs to generate complex B*. Complex C is responsible for the two transesterification reactions at the SS. Additional rearrangements result in the excision of the intron, which is removed as a lariat RNA, and ligation of the exons. The U2, U5, and U6 snRNPs are then released from the complex and recycled for subsequent rounds of splicing.
Figure from: Suñé-Pou, M., et. al. (2017) Genes 8(3):87
In mammals, the first catalytic step of the splicing reaction begins when the U1 snRNP binds the 5′ SS of the intron (defined by the consensus sequence AGGURAGU), and the splicing factors SF1 and U2AF cooperatively recognize the BPS, Py, and 3′ SS to assembled complex E or the commitment complex (Figure 10.23). Subsequently, U2 snRNP and additional proteins are recruited to the pre-mRNA BPS to form the pre-spliceosome or complex A. The binding of the U4/U6-U5 tri-snRNP forms the pre-catalytic spliceosome or complex B. After RNA-RNA and RNA-protein rearrangements at the heart of the spliceosome, U1 and U4 are released to form the activated complex B or complex B* This complex is responsible for executing the first catalytic step, through which the phosphodiester bond at the 5′ SS of the intron is modified by the 2′-hydroxyl of an adenosine of the BPS to form a free 5′ exon and a branched intron (Fig. 10.24). The reaction of the 2′-hydroxyl from the branchpoint adenosine nucleotide is known as a transesterification reaction. During this process, additional rearrangements occur to generate the catalytic spliceosome or complex C (Fig. 10.23), which is responsible for catalyzing the second transesterification reaction leading to intron excision and exon–exon ligation (Fig. 10.24). The resulting intron structure is referred to as a lariat structure. After the second catalytic step, the U2, U5, and U6 snRNPs are released from the post-spliceosomal complex and recycled for additional rounds of splicing.
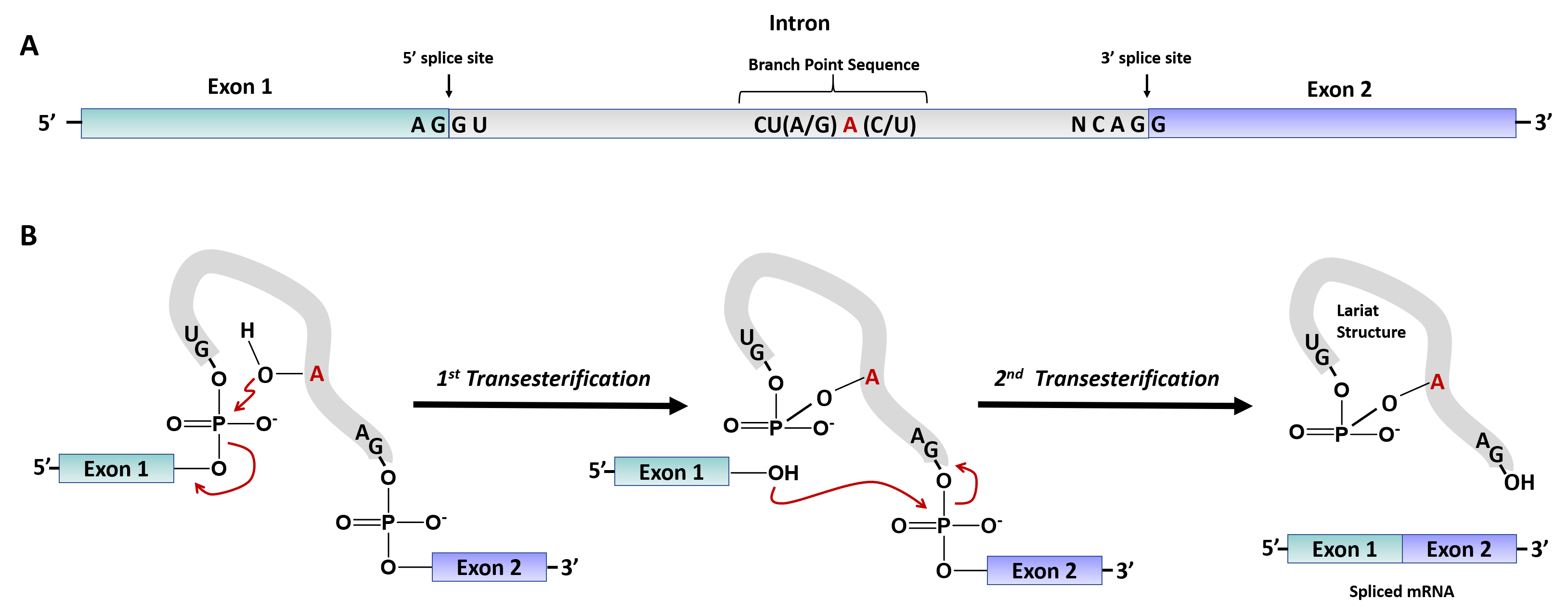
Figure 10.24 Transesterification Reactions Involved in mRNA Splicing. (A) Schematic diagram of the pre-mRNA with exons and introns indicated. Key sequences are required for splicing at the 5′ and 3′ intron locations, and for the recognition and positioning of the branchpoint Adenosine residue for the first transesterification reaction. (B) Schematic of the two transesterification reactions required for intron removal. The branchpoint 2′-OH residue mediates attack on the 5′-phosphate of the intron guanosine residue located at the 5′-splice site. This releases the 3′ hydroxyl of Exon 1 which subsequently mediates attack of the 5′ phosphate of the first guanosine residue in Exon 2. The 3′ hydroxyl of the intron guanine residue is released forming the Lariat structure and Exon 1 is ligated to Exon 2.
Alternative Splicing (AS) offers an additional mechanism for regulating protein production and function. AS options are determined by the expression of or exposure to in trans elements present within unique cellular locations and environments. Additional sequence elements within the mRNA, known as exonic and intronic splicing silencers or enhancers (ESS, ISS, ESE, and ISE, respectively), participate in the regulation of AS. Specific RNA-binding proteins, including heterogeneous nuclear ribonucleoproteins (hnRNPs) and serine/arginine-rich (SR) proteins, recognize these sequences to positively or negatively regulate AS (Figure 10.25). These regulators, together with an ever-increasing number of additional auxiliary factors, provide the basis for the specificity of this pre-mRNA processing event in different cellular locations within the body.
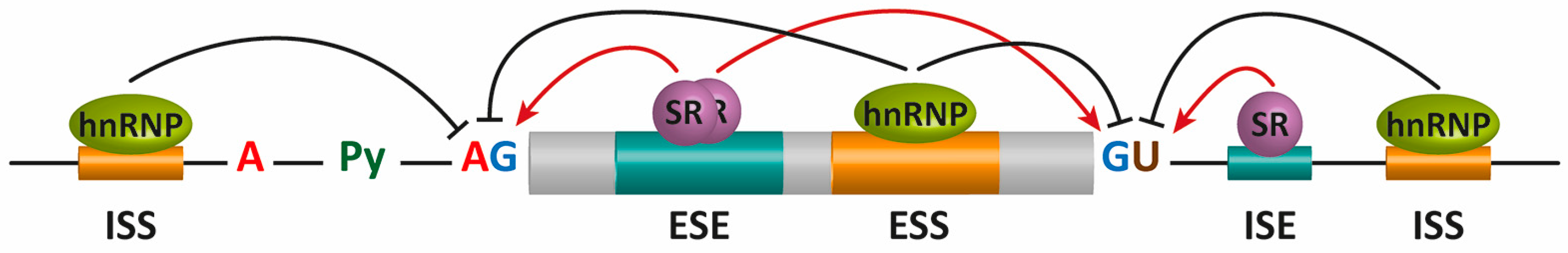
Figure 10.25 Alternative splicing (AS) regulation by cis mRNA elements and trans-acting factors. The core cis sequence elements that define the exon/intron boundaries (5′ and 3′ splice sites (SS), GU-AG, polypyrimidine (Py) tract, and branch point sequence (BPS)) are poorly conserved. Additional enhancer and silencer elements in exons and in introns (ESE: exonic splicing enhancers; ESI: exonic splicing silencers; ISE: intronic splicing enhancers; ISI: intronic splicing silencers) contribute to the specificity of AS regulation. Trans-acting splicing factors, such as SR family proteins and heterogeneous nuclear ribonucleoprotein particles (hnRNPs), bind to enhancers and silencers and interact with spliceosomal components. In general, SR proteins bound to enhancers facilitate exon definition, and hnRNPs inhibit this process. These trans-acting elements are expressed differentially within different locations or under different environmental stimuli to regulate AS.
Figure from: Suñé-Pou, M., et. al. (2017) Genes 8(3):87
There are several different types of AS events, which can be classified into four main subgroups. The first type is exon skipping, which is the major AS event in higher eukaryotes. In this type of event, a cassette exon is removed from the pre-mRNA (Fig. 10.26 a). The second and third types are alternative 3′ and 5′ SS selection (Fig. 10.26 b & c). These types of AS events occur when the spliceosome recognizes two or more splice sites at one end of an exon. The fourth type is intron retention (Fig. 10.26 d), in which an intron remains in the mature mRNA transcript. This AS event is much more common in plants, fungi and protozoa than in vertebrates. Other events that affect the transcript isoform outcome include mutually exclusive exons (Fig. 10.26 e), alternative promoter usage (Fig. 10.26 f), and alternative polyadenylation (Fig. 10.26 g).
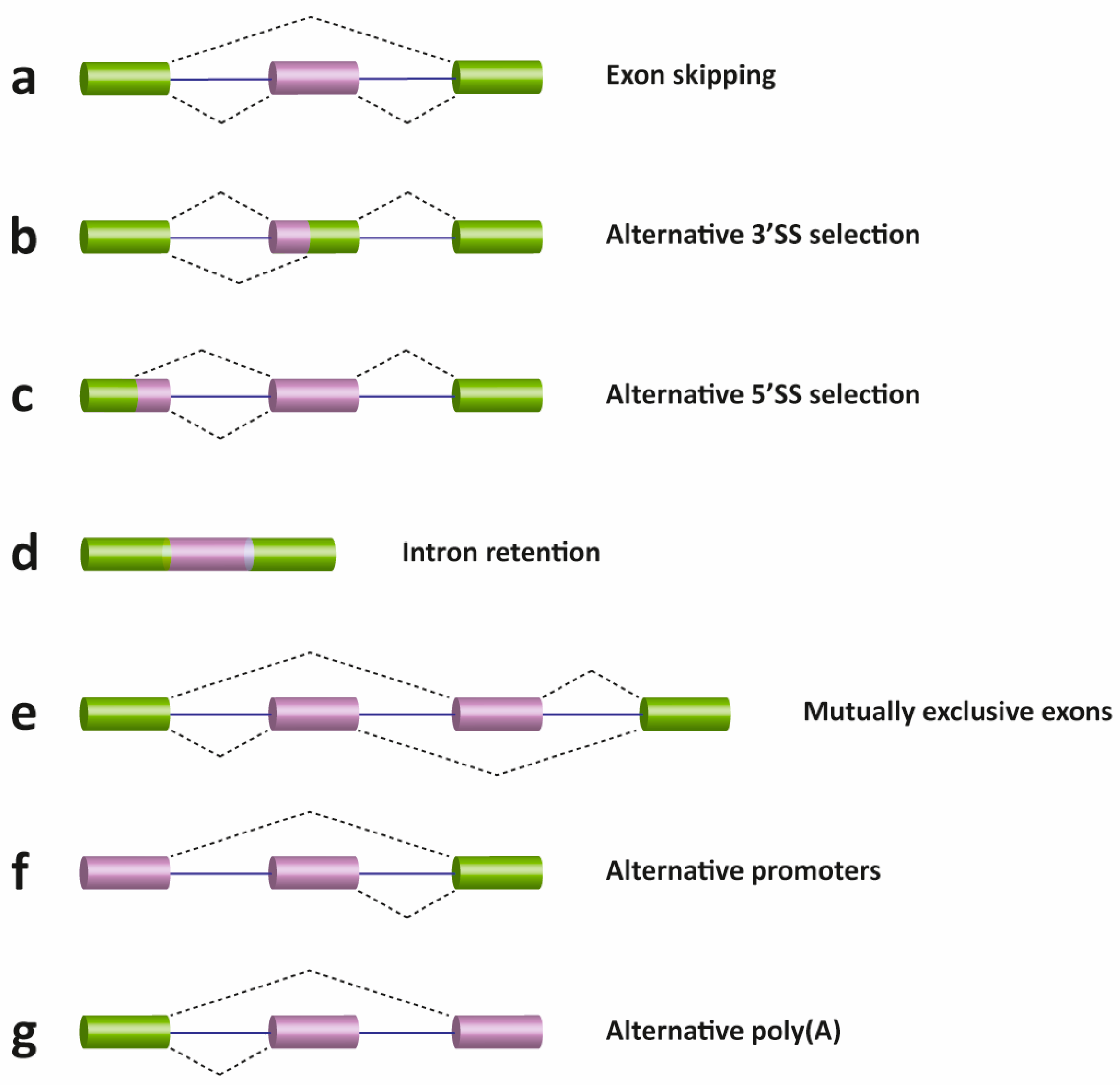
Figure 10.26 Schematic representation of different types of alternative transcriptional or splicing events, with exons (boxes) and introns (lines). Constitutive exons are shown in green and alternatively spliced exons in purple. Dashed lines indicate the AS event. Exon skipping (a); alternative 3′ (b) and 5′ SS selection (c); intron retention (d); mutually exclusive exons (e); alternative promoter usage (f); and alternative polyadenylation (g) events are shown. Like alternative splicing (AS), usage of alternative promoter and polyadenylation sites allow a single gene to encode multiple mRNA transcripts.
Figure from: Suñé-Pou, M., et. al. (2017) Genes 8(3):87
Back to the Top
10.6 References
- Parker, N., Schneegurt, M., Thi Tu, A-H., Lister, P., Forster, B.M. (2019) Microbiology. Openstax. Available at: https://opentextbc.ca/microbiologyopenstax/
- Palazzo, A., and Lee, E.S. (2015) Non-coding RNA: what is function and what is junk? Frontiers in Genetics 6:2 Available at: file:///C:/Users/flatt/AppData/Local/Temp/fgene-06-00002.pdf
- Wikipedia contributors. (2020, July 9). RNA. In Wikipedia, The Free Encyclopedia. Retrieved 15:30, August 6, 2020, from https://en.wikipedia.org/w/index.php?title=RNA&oldid=966784317
- Burenina, O.Y., Oretskaya, T.S., and Kubareva, E.A. (2017) Non-Coding RNAs As Transcriptional Regulators in Eukaryotes. Acta Naturae 9(4):13-25. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5762824/
- Khatter, H., Vorlander, M.K., and Muller C.W. (2017) RNA polymerase I and III: similar yet unique. Current Opinion in Structural Biology 47:88-94. Available at: https://www.sciencedirect.com/science/article/pii/S0959440X17300313
- Wikipedia contributors. (2020, May 8). Sigma factor. In Wikipedia, The Free Encyclopedia. Retrieved 17:50, August 7, 2020, from https://en.wikipedia.org/w/index.php?title=Sigma_factor&oldid=955570499
- Bae, B., Felkistov, A., Lass-Napiokowska, A., Landick, R., and Darst, S.A. (2015) Structure of a bacterial RNA polymerase holoenzyme open protomer complex. eLife 4:e08504. Available at: https://elifesciences.org/articles/08504
- Petrenko, N., Jin, Y., Dong, L., Wong, K.H., and Struhl, K. (2019) Requirements for RNA polymerase II preinitiation complex formation in vivo. eLife 8:e43654. Available at: https://elifesciences.org/articles/43654
- Gupta, K., Sari-Ak, D., Haffke, M., Trowitzsch, S., and Berger, I. (2016) Zooming in on transcription preinitiation. J Mol Biol. 428(12):2581-2591. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4906157/
- Wikipedia contributors. (2020, April 17). TATA-binding protein. In Wikipedia, The Free Encyclopedia. Retrieved 14:54, August 8, 2020, from https://en.wikipedia.org/w/index.php?title=TATA-binding_protein&oldid=951583592
- Patel, A.B., Greber, B.J., and Nogales, E. (2020) Recent insights into the structure of TFIID, its assembly, and its binding to core promoter. Curr Op Struct Bio 61:17-24. Available at: https://www.sciencedirect.com/science/article/pii/S0959440X19301113#fig0010
- Ruff, E.F., Record, Jr., M.T., Artsimovitch, I., (2015) Initial events in bacterial transcription initiation. Biomolecules 5(2):1035-1062. Available at: https://www.mdpi.com/2218-273X/5/2/1035/htm
- Kireeva, M., Opron, K., Seibold, S., Domecq, C., Cukier, R.I., Coulombe, B., Kashlev, M., and Burton, Z. (2102) Molecular dynamics and mutational analysis of the catalytic and translocation cycle of RNA polymerase. BMC Biophysics 5(1):11. Available at: https://www.researchgate.net/publication/225281979_Molecular_dynamics_and_mutational_analysis_of_the_catalytic_and_translocation_cycle_of_RNA_polymerase/figures?lo=1
- Washburn, R.S., and Gottesman, M.E. (2015) Regulation of transcription elongation and termination. Biomolecules 5(2):1063-1078. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4496710/pdf/biomolecules-05-01063.pdf
- Zenkin, N., and Yuzenkova, Y. (2015) New insights into the functions of transcription factors that bind the RNA polymerase secondary channel. Biomolecules 5(3):1195-1209. Available at: https://www.mdpi.com/2218-273X/5/3/1195/htm
- Gocheva, V., LeGall, A., Boudvillain, M., Margeat, E., and Nollmann, M. (2015) Direct observation of the translocation mechanism of transcription termination factor Rho. Nuc Acids Res 43(1):10.1093. Available at: https://www.researchgate.net/publication/272162172_Direct_observation_of_the_translocation_mechanism_of_transcription_termination_factor_Rho
- Miki, T.S., Carl, S.H., and Groβhans, H. (2017) Two disctinct transcription termination modes dictated by promoters. Genes & Dev 31:1-10. Available at: https://www.researchgate.net/publication/320350041_Two_distinct_transcription_termination_modes_dictated_by_promoters
- Gurumurthy, A., Shen, Y., Gunn, E.M., Bungert, J. (2018) Phase separation and transcription regulation: Are Super-Enhancers and Locus Control Regions primary sites of transcription complex assembly? BioEssays 1800164. Available at: https://www.researchgate.net/publication/329331157_Phase_Separation_and_Transcription_Regulation_Are_Super-Enhancers_and_Locus_Control_Regions_Primary_Sites_of_Transcription_Complex_Assembly
- Suñé-Pou, M., Prieto-Sánchez, Boyero-Corral, S., Moreno-Castro, C., El Yousfi, Y., Suñé-Negre, J.M., Hernández-Munain, C., and Suñé, C. (2017) Targeting splicing in the treatment of human disease. Genes 8(3):87. Available at: https://www.mdpi.com/2073-4425/8/3/87/htm
- Schaughency, P., Merran, J., and Corden J.L. (2014) Genome-wide mapping of yeast RNA polymerase II termination. PLOS Genetics 10(10):e1004632 Available at: https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1004632
- Nourse, J., Spada, S., and Danckwardt, S. (2020) Emerging roles of RNA 3′-end cleavage and polyadenylation in pathogenesis, diagnosis, and therapy of human disorders. Biomolecules 10(6):915. Available at: https://www.mdpi.com/2218-273X/10/6/915/htm
- Wikipedia contributors. (2020, July 30). Five-prime cap. In Wikipedia, The Free Encyclopedia. Retrieved 05:53, August 11, 2020, from https://en.wikipedia.org/w/index.php?title=Five-prime_cap&oldid=970240533
- Cortes, T. and Cox, R.A. (2015) Transcription and translation of the rpsJ, rplN and rRNA operons of the tubercle bacillus. Microbiology (2015) 161:719-728. Available at: https://www.microbiologyresearch.org/docserver/fulltext/micro/161/4/719_mic000037.pdf?expires=1597159574&id=id&accname=guest&checksum=6FFC9C066EF41C7799FAE843CE94C49F
- Hein, P.P. and Landick, R. (2010) The bridge helix coordinates movements of modules in RNA polymerase. BMC Biology 8:141. Available at: https://bmcbiol.biomedcentral.com/articles/10.1186/1741-7007-8-141
- Gonatopoulos-Pournatzis, T., and Cowling, V.H. (2014) Cap-binding complex (CBC). Biochem. J. 457:231-242. Available at: https://www.researchgate.net/publication/259392894_Cap-binding_complex_CBC