Home » Student Resources » Online Chemistry Textbooks » CH450 and CH451: Biochemistry - Defining Life at the Molecular Level » Chapter 12: DNA Damage and Repair
MenuCH450 and CH451: Biochemistry - Defining Life at the Molecular Level
Chapter 12: DNA Damage and Repair
12.1 DNA Mutations
12.2 Types of DNA Damage
12.3 Cellular Stress and DNA Damage Response
12.4 Mismatch Repair
12.5 Base Excision Repair
12.6 Nucleotide Excision Repair
12.7 Repair of Double-Stranded DNA breaks
12.8 Error-Prone Bypass and Translesion Synthesis
12.9 Practice Problems
12.10 References
12.1 DNA Mutations
The integrity of the DNA structure for cell viability is underscored by the vast amounts of cellular machinery dedicated to ensure its accurate replication, repair, and storage. Even still, mutations within the DNA are a fairly common event.
Mutations are random changes that occur within the sequence of bases in DNA. They can be large scale, altering the structure of the chromosomes, or small scale where they only alter a few or even a single base or nucleotide. Mutations can occur for many reasons. For example, DNA mutations can be caused by mistakes made by the DNA polymerase during replication. As noted in chapter 9, DNA polymerases are highly processive enzymes that contain proofreading and editing functions. With these safeguards, their error rates are typically very low and range from one in a million bases to one in a billion bases. Even with such high fidelity, this error rate will lead to between 3 and 3,000 errors within the human genome for each cell undergoing DNA replication. DNA mutations can also result through the replication of DNA that has been damaged by endogenous or exogenous agents. The next section will highlight common types of DNA damage and their effects. If a DNA polymerase encounters a damaged DNA base in the template DNA during replication it may place a random nucleotide base across from the lesion. For example, an adenine-containing nucleotide will often be added across a lesion, regardless of what the correct match should be. This can lead to the formation of transition or transversion mutations.
A transition mutation is a point mutation that changes a purine nucleotide to another purine (A ↔ G) or a pyrimidine nucleotide to another pyrimidine (C ↔ T). Whereas, a transversion refers to the substitution of a purine for a pyrimidine or vice versa. Sometimes lesions may cause bases to be skipped during replication or cause extra nucleotides to be inserted into the backbone. DNA polymerases can also slip during the replication of regions of the DNA that have repeated sequences or large stretches repeating a single base. Larger lesions or cross-links in the DNA during replication can lead to more catastrophic DNA damage including DNA strand breaks. Mutations may also occur during the processes of mitosis and meiosis when sister chromatids and/or homologous chromosomes are being separated from one another.
In nature, mutagenesis, or the process of generating DNA mutations, can lead to changes that are harmful, or beneficial, or have no effect. Harmful mutations can lead to cancer and various heritable diseases, but beneficial mutations are the driving force of evolution. In 1927, Hermann Muller first demonstrated the effects of mutations with observable changes in chromosomes. He induced mutagenesis by irradiating fruit flies with X-rays.
When a mutation is caused by an environmental factor or a chemical agent, that agent is called a mutagen. Typical mutagens include chemicals, like those inhaled while smoking, and radiation, such as X-rays, ultraviolet light, and nuclear radiation. Different mutagens have a different modes of damaging DNA and are discussed further in the next section. It is important to note that DNA damage, in and of itself, does not necessarily lead to the formation of a mutation in the DNA. There are elaborate DNA repair processes designed to recognize and repair different types of DNA lesions. Fewer than 1 in 1,000 DNA lesions will actually result in a DNA mutation. The processes of DNA damage recognition and repair are the focus of later sections within this chapter.
Types of Mutations
There are a variety of types of mutations. Two major categories of mutations are germline mutations and somatic mutations.
- Germline mutations occur in gametes, the sex cells, such as eggs and sperm. These mutations are especially significant because they can be transmitted to offspring and every cell in the offspring will have the mutations.
- Somatic mutations occur in other cells of the body. These mutations may have little effect on the organism because they are confined to just one cell and its daughter cells. Somatic mutations also cannot be passed on to offspring.
Mutations also differ in the way that the genetic material is changed. Mutations may change an entire chromosome or just one or a few nucleotides.
Chromosomal alterations are mutations that change chromosome structure or number. They occur when a section of a chromosome breaks off and rejoins incorrectly or does not rejoin at all. Possible ways these mutations can occur are illustrated in the figure below. Chromosomal alterations are very serious. They often result in the death of the cell or organism in which they occur. If the organism survives, it may be affected in multiple ways. An example of a human chromosomal alteration is the mutation that causes Down Syndrome. It is a duplication mutation that leads to developmental delays and other abnormalities. It occurs when the individual inherits an extra copy of chromosome 21. It is also called trisomy (“three-chromosome”) 21. Thus, large-scale mutations in chromosomal structure include: (1) Amplifications (including gene duplications) where repetition of a chromosomal segment or presence of extra piece of a chromosome broken piece of a chromosome may become attached to a homologous or non-homologous chromosome so that some of the genes are present in more than two doses leading to multiple copies of all chromosomal regions, increasing the dosage of the genes located within them, (2) Deletions of large chromosomal regions, leading to loss of the genes within those regions, and (3) Chromosomal Rearrangements such as translocations (which interchange of genetic parts from nonhomologous chromosomes), insertions (which insert segments of one chromosome into another nonhomologous chromosome), and inversions (which invert or flip a section of a chromosome into the opposite orientation)(Figure 12.1).
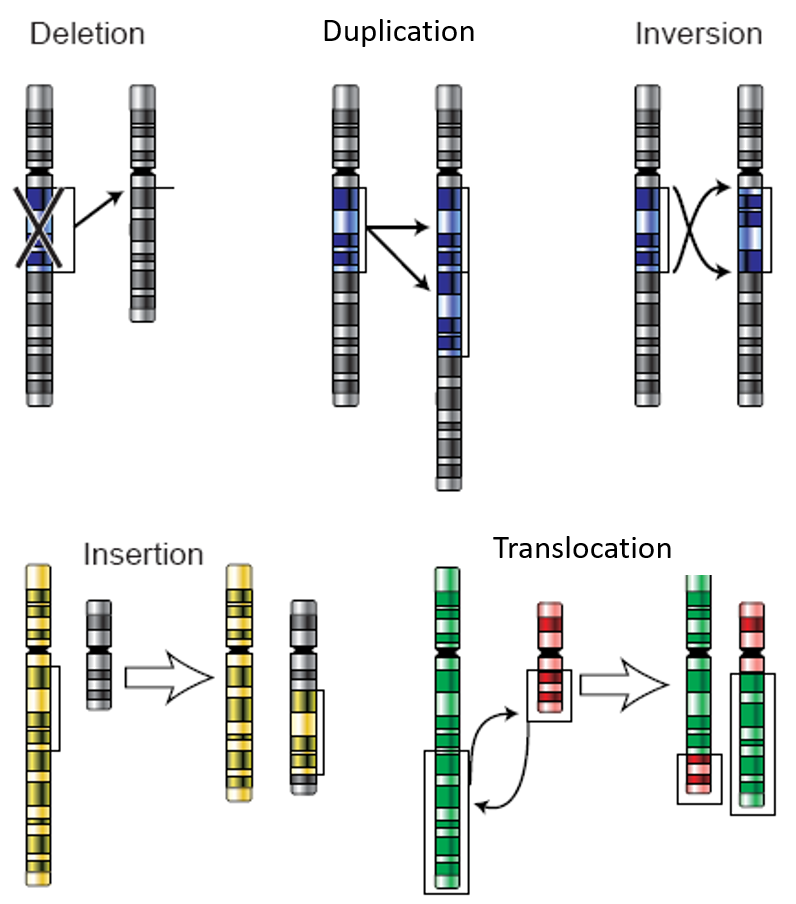
Figure 12.1 Chromosomal Alterations. Chromosomal alterations are major changes in the genetic material.Image modified from Dietzel65
There are also smaller mutations that can occur that only alter a single nucleotide or a small number of nucleotides within a localized region of the DNA. These are classified according to how the DNA molecule is altered. One type, a point mutation, affects a single base and most commonly occurs when one base is substituted or replaced by another. Mutations also result from the addition of one or more bases, known as an insertion, or the removal of one or more bases, known as a deletion.
Point mutations may have a wide range of effects on protein function (Table 12.1 and Figure 12.2). As a consequence of the degeneracy of the genetic code, a point mutation will commonly result in the same amino acid being incorporated into the resulting polypeptide despite the sequence change. This change would have no effect on the protein’s structure, and is thus called a silent mutation. A missense mutation results in a different amino acid being incorporated into the resulting polypeptide. The effect of a missense mutation depends on how chemically different the new amino acid is from the wild-type amino acid. The location of the changed amino acid within the protein also is important. For example, if the changed amino acid is part of the enzyme’s active site or greatly affects the shape of the enzyme, then the effect of the missense mutation may be significant. Many missense mutations result in proteins that are still functional, at least to some degree. Sometimes the effects of missense mutations may be only apparent under certain environmental conditions; such missense mutations are called conditional mutations. Rarely, a missense mutation may be beneficial. Under the right environmental conditions, this type of mutation may give the organism that harbors it a selective advantage. Yet another type of point mutation, called a nonsense mutation, converts a codon encoding an amino acid (a sense codon) into a stop codon (a nonsense codon). Nonsense mutations result in the synthesis of proteins that are shorter than the wild type and typically not functional (Summarized in Figure 12.3).
Table 12.1: Types of Point Mutations
| Type | Description | Example | Effect |
|---|---|---|---|
| Silent | mutated codon codes for the same amino acid | CAA (glutamine) → CAG (glutamine) | none |
| Missense | mutated codon codes for a different amino acid | CAA (glutamine) → CCA (proline) | variable |
| Nonsense | a mutated codon is a premature stop codon | CAA (glutamine) → UAA (stop) usually | serious |

Figure 12.2 The Potential Effects of Point Mutations on Protein Coding Regions. The image shows various types of point mutations (silent, missense, and nonsense), which may lead to change in the protein structure.
Figure from: Jonsta247
Smaller scale deletions and insertions also cause various effects. Because codons are triplets of nucleotides, insertions or deletions in groups of three nucleotides may lead to the insertion or deletion of one or more amino acids and may not cause significant effects on the resulting protein’s functionality. However, frameshift mutations, caused by insertions or deletions of a number of nucleotides that are not a multiple of three are extremely problematic because a shift in the reading frame results (Figure 12.3). Because ribosomes read the mRNA in triplet codons, frameshift mutations can change every amino acid after the point of the mutation. The new reading frame may also include a stop codon before the end of the coding sequence. Consequently, proteins made from genes containing frameshift mutations are nearly always nonfunctional.
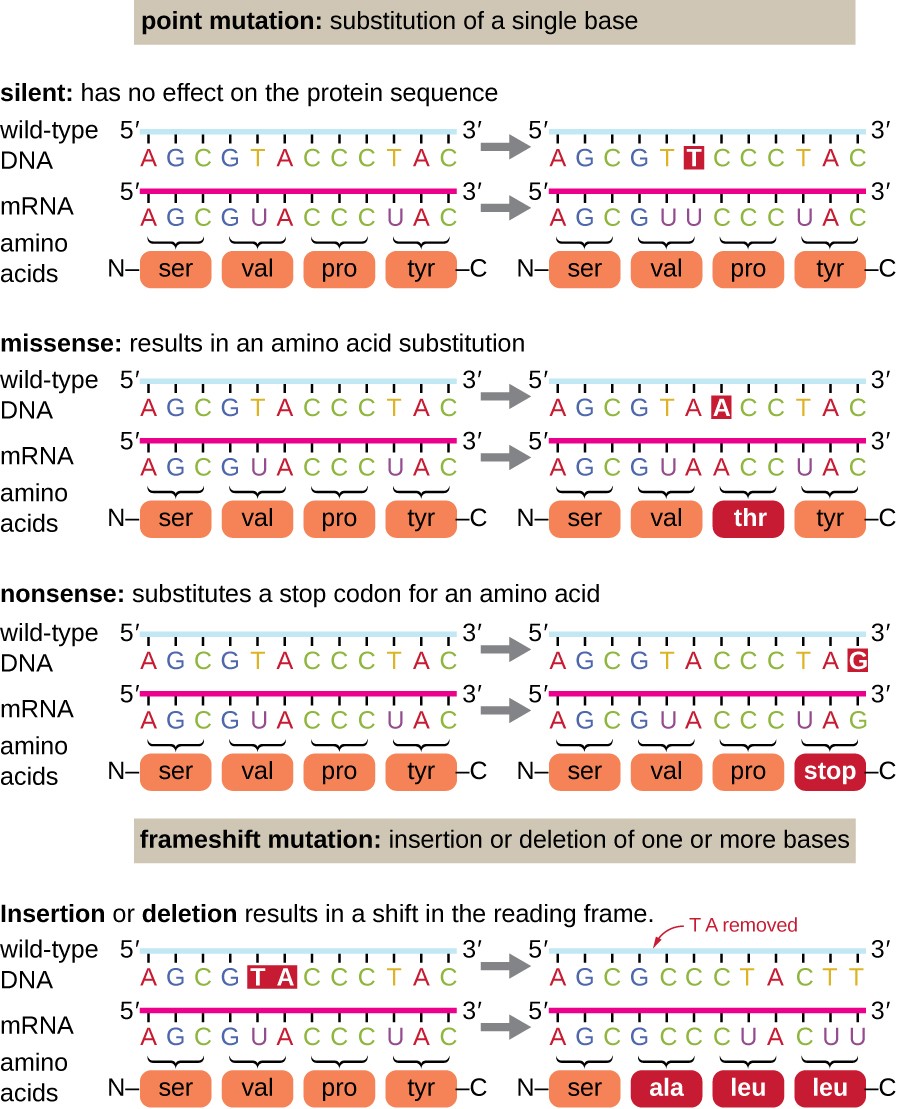
Figure 12.3. Summary of Small Scale Mutations on Protein Coding Regions. DNA alterations that lead to changes in the protein sequence encoded by the DNA can include point mutations, DNA insertions, and DNA deletions.
Figure from: Parker, et al (2019) Microbiology from Openstax
The majority of mutations have neither negative nor positive effects on the organism in which they occur. These mutations are called neutral mutations. Examples include silent point mutations, which are neutral because they do not change the amino acids in the proteins they encode.
Some mutations have a positive effect on the organism in which they occur. They are referred to as beneficial mutations. If they occur in germline cells (eggs or sperm) these traits can be heritable and passed from one generation to the next. Beneficial mutations generally code for new versions of proteins that help organisms adapt to their environment. If they increase an organism’s chances of surviving or reproducing, the mutations are likely to become more common within a population over time. There are several well-known examples of beneficial mutations. Here are just two:
- Mutations have occurred in bacteria that allow the bacteria to survive in the presence of antibiotic drugs. The mutations have led to the evolution of antibiotic-resistant strains of bacteria.
- A unique mutation is found in people in a small town in Italy. The mutation protects them from developing atherosclerosis, which is the dangerous buildup of fatty materials in blood vessels. The individual in which the mutation first appeared has even been identified.
Harmful mutations can also occur. Imagine making a random change in a complicated machine such as a car engine. The chance that the random change would improve the functioning of the car is very small. The change is far more likely to result in a car that does not run well or perhaps does not run at all. By the same token, any random change in a gene’s DNA is more likely to result in the production of a protein that does not function normally or may not function at all, than in a mutation that improves the function. Such mutations are likely to be harmful. Harmful mutations may cause genetic disorders or cancer.
- A genetic disorder is a disease, syndrome, or other abnormal condition caused by a mutation in one or more genes or by a chromosomal alteration. An example of a genetic disorder is cystic fibrosis. A mutation in a single gene causes the body to produce thick, sticky mucus that clogs the lungs and blocks ducts in digestive organs. Genetic disorders are usually caused by gene mutations that occur within germline cells and are heritable in nature.
- Illnesses caused by mutations that occur within an individual, but are not passed on to their offspring, are mutations that occur in somatic cells. Cancer is a disease caused by an accumulation of mutations within somatic cells. It results in cells that grow out of control and form abnormal masses of cells called tumors. It is generally caused by mutations in genes that regulate the cell cycle, DNA repair, angiogenesis, and other genes that favor cell growth and survival. Because of the mutations, cells with the mutated DNA have evolved to divide without restrictions, hide from the immune system, and develop drug resistance.
Back to the Top
12.2 Types of DNA Damage
DNA damage, due to environmental factors and normal metabolic processes inside the cell, occurs at a rate of 1,000 to 1,000,000 molecular lesions per cell per day. While this constitutes only 0.000165% of the human genome’s approximately 6 billion bases (3 billion base pairs), if left unrepaired can cause mutations in critical genes (such as tumor suppressor genes) can impede a cell’s ability to carry out its function and appreciably increase the likelihood of tumor formation and disease states such as cancer.
The vast majority of DNA damage affects the primary structure of the double helix; that is, the bases themselves are chemically modified. These modifications can, in turn, disrupt the molecules’ regular helical structure by introducing non-native chemical bonds or bulky adducts that do not fit in the standard double helix. Unlike proteins and RNA, DNA usually lacks tertiary structure and therefore damage or disturbance does not occur at that level. DNA is, however, supercoiled and wound around “packaging” proteins called histones (in eukaryotes), and both superstructures are vulnerable to the effects of DNA damage.
There are several types of DNA damage that can occur due either to normal cellular processes or due to the environmental exposure of cells to DNA damaging agents. DNA bases can be damaged by: (1) oxidative processes, (2) alkylation of bases, (3) base loss caused by the hydrolysis of bases, (4) bulky adduct formation, (5) DNA crosslinking, and (6) DNA strand breaks, including single and double stranded breaks. An overview of these types of damage are described below.
Oxidative Damage
Reactive oxygen species (ROS) can cause significant cellular stress and damage including the oxidative damage of DNA. Hydroxyl radicals (•OH) are one of the most reactive and electrophilic of the ROS and can be produced by ultraviolet and ionizing radiations or from other radicals arising from enzymatic reactions. The •OH can cause the formation of 8-oxo-7,8-dihydroguanine (8-oxoG) from guanine residues, among other oxidative products (Figure 12.4). Guanine is the most easily oxidized of the nucleic acid bases, because it has the lowest ionization potential among the DNA bases. The 8-oxo-dG is one of the most abundant DNA lesions, and it considered as a biomarker of oxidative stress. It has been estimated that up to 100,000 8-oxo-dG lesions can occur daily in DNA per cell. The reduction potential of 8-oxo-dG is even lower (0.74 V vs. NHE) than that of guanosine (1.29 V vs NHE). Therefore, it can be further oxidized creating a variety of secondary oxidation products.

Figure 12.4 Reactive Oxygen Species and DNA Damage. Under conditions of oxidative stress, 8-oxoG is the result of reactive oxygen species (ROS) modifying a guanine.
Figure from: Poetsch, A.R. (2020) Comp & Struct Biotech J. 18:207-219.
As mentioned previously, increased levels of 8-oxo-dG in a tissue can serve as a biomarker of oxidative stress. Furthermore, increased levels of 8-oxo-dG are frequently found associated with carcinogenesis and other disease states (Figure 12.5). During the replication of DNA that contains 8-oxo-dG, adenine is most often incorporated across from the lesion. Following replication, the 8-oxo-dG is excised during the repair process and a thymine is incorporated in its place. Thus, 8-oxo-dG mutations typically result in a G to T transversion.
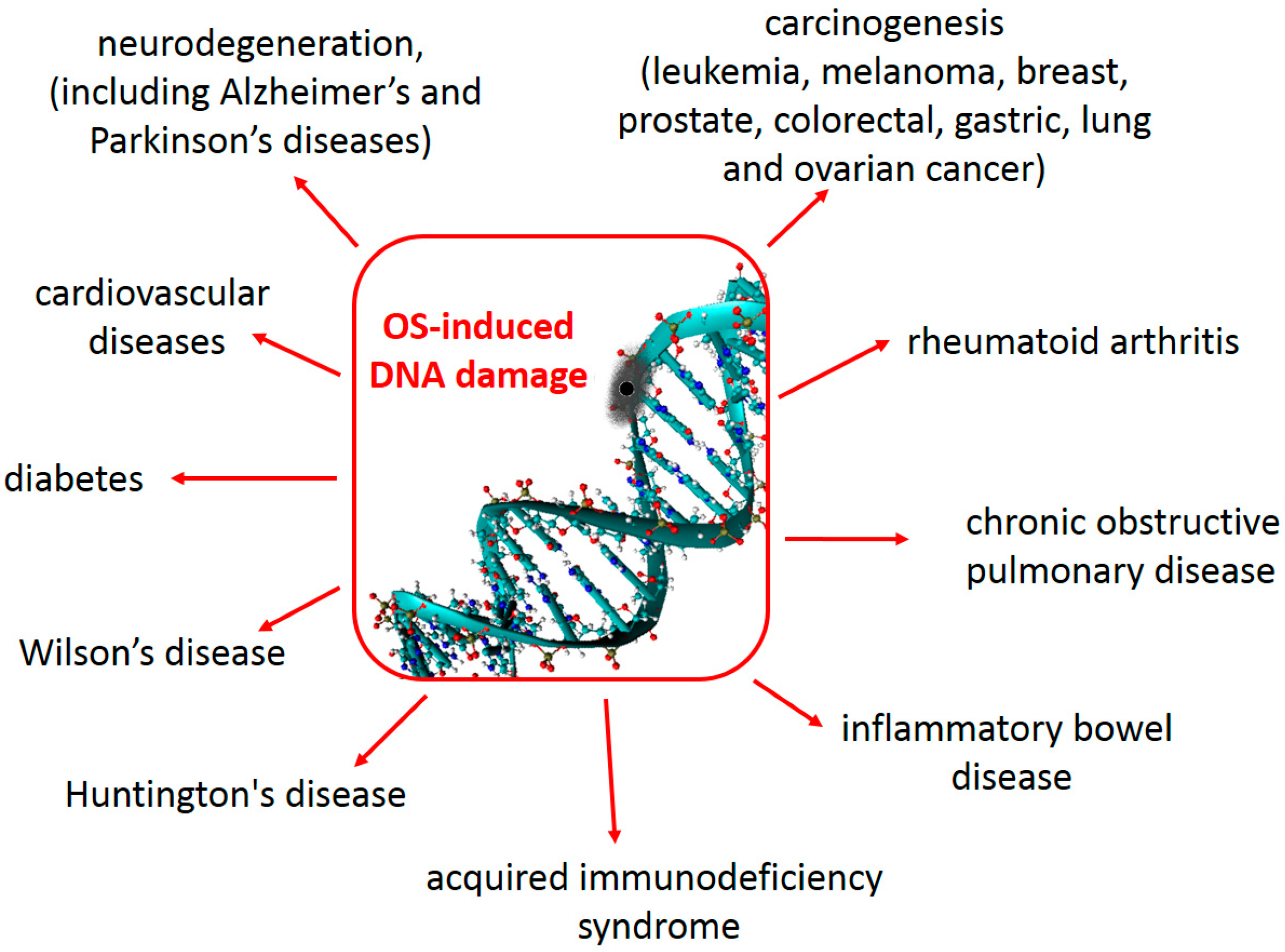
Figure 12.5 Oxidative Stress and Human Health. DNA damage caused by oxidative stress has been associated with many human disease states.
Figure from: Galano, A., Tan, D-X., and Reiter, R.J. (2018) Molecules 23(3)530
Alkylation of Bases
Alkylating agents are widespread in the environment and are also produced endogenously, as by-products of cellular metabolism. They introduce lesions into DNA or RNA bases that can be cytotoxic, mutagenic, or neutral to the cell. Figure 12.6 depicts the major reactive sites on the DNA bases that are susceptible to alkylation. Cytotoxic lesions block replication, interrupt transcription, or signal the activation of apoptosis, whereas mutagenic ones are miscoding and cause mutations in newly synthesized DNA.The most common type of alkylation is methylaton with the major products including N7-methylguanine (7meG), N3-methyladenine (3meA), and O6-methylguanine (O6meG). Smaller amounts methylation also occurs on other DNA bases, and include the formation of N1-methyladenine (1meA), N3-methylcytosine (3meC), O4-methylthymine (O4meT), and methyl phosphotriesters (MPT).
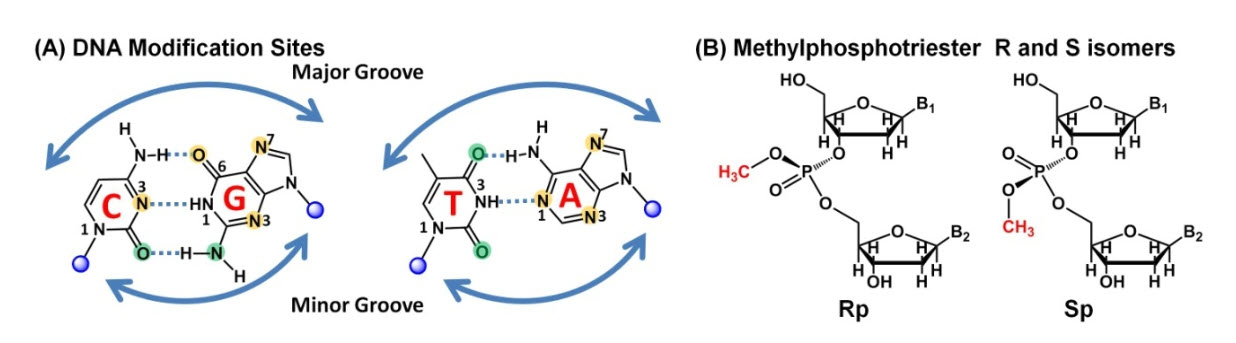
Figure 12.6 Major Sites of Alkylation in DNA. The diagram shows DNA Watson-Crick base pairs with principal damage sites modified by small alkylating agents (methyl and ethyl). (A) Sites of base modification by SN1 and SN2 alkylating agents. The orange indicates major damage sites andthe green minor damage sites. (B) Phosphotriester formation indicated by the presence of methyl groups, along withthe Rp and Sp isomers.
Figure from: Ahmad, A., Nay, S.L. and O’Conner, T.R. (2015) Chapter 4 Direct Reversal Repair in Mammalian Cells
Alkylating agents can cause damage at all exocyclic nitrogens and oxygens in DNA and RNA, as well as at ring nitrogens (Figure 12.6A). However, the percentage of each base site modified depends on the alkylating agent, the position in DNA or RNA, and whether nucleic acids are single- or double-stranded. Interestingly, O-alkylations are more mutagenic and harmful than N-alkylations, which may be more cytotoxic, but not as mutagenic.
As we will explore in Chapter 13, methylation of DNA also serves as an important mechanism regulating gene expression.
Base Loss
An AP site (apurinic/apyrimidinic site), also known as an abasic site, is a location in DNA (also in RNA but much less likely) that has neither a purine nor a pyrimidine base, either spontaneously or due to DNA damage (Figure 12.7). It has been estimated that under physiological conditions 10,000 apurinic sites and 500 apyrimidinic may be generated in a cell daily.

Figure 12.7 Abasic Sites. Apurinic and Apyrimidimic (AP) sites occur due to unstable hydrolysis.
Figure from Mayhew
AP sites can be formed by spontaneous depurination, but also occur as intermediates in base excision repair, the repair process described in section 12.5. If left unrepaired, AP sites can lead to mutation during semiconservative replication. They can cause replication fork stalling and are often bypassed by translesion synthesis, that is discussed in greater detail in section 12.8. In E. coli, adenine is preferentially inserted across from AP sites, known as the “A rule”. The situation is more complex in higher eukaryotes, with different nucleotides showing a preference depending on the organism and environmental conditions.
Bulky Adduct Formation
Some chemicals are biologically reactive and will form covalent linkages with biological molecules such as DNA and proteins creating large bulky adducts, or appendages, that branch off from the main molecule. We will use the mutagen/carcinogen, benzo[a]pyrene, as an example for this process.
Benzo[a]pyrene is a polycyclic aromatic hydrocarbon that forms during the incomplete combustion of organic matter at temperatures between 300°C (572°F) and 600°C (1,112°F). The ubiquitous compound can be found in coal tar, tobacco smoke and many foods, especially grilled meats. Benzo[a]pyrene is actually a procarcinogen that needs to be biologically activated by metabolism before it forms a reactive metabolite (Figure 12.8) Normally, when the body is exposed to foreign molecules, it will start a metabolic process that makes the molecule more hydrophilic and easier to remove as a waste product. Unfortunately, in the case of benzo[a]pyrene, the resulting metabolite is a highly reactive epoxide that forms a bulky adduct preferentially with guanine residues in DNA. If left unrepaired, during DNA replication an adenine will usually be placed across from the lesion in the daughter molecule. Subsequent repair of the adduct will result in the replacement of the damaged guanine base with a thymine, causing a G –> T transversion mutation.
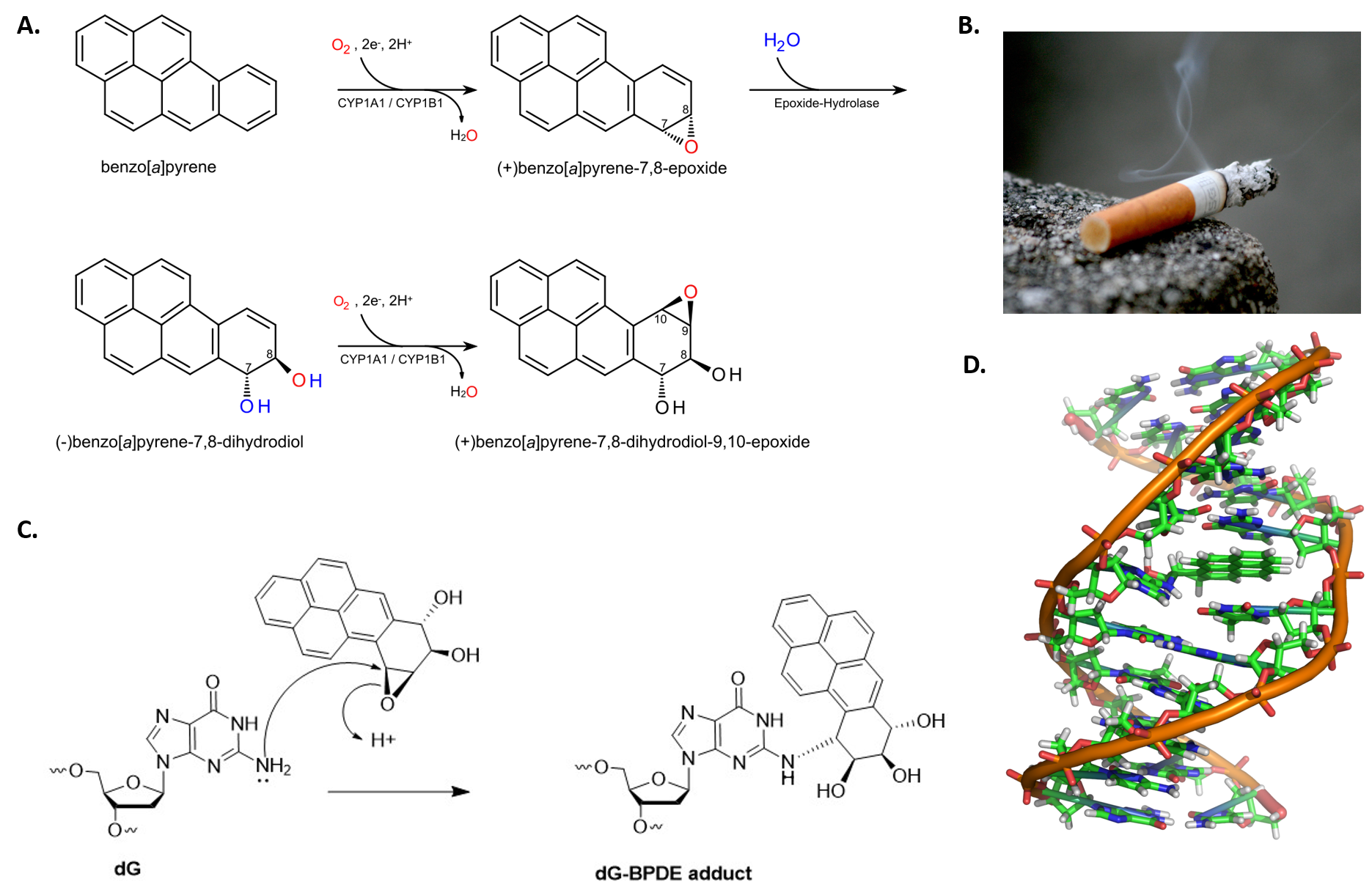
Figure 12.8 Benzo[a]pyrene Activation and Adduct Formation. (A) Benzo[a]pyrene is biologically activated to a dihydrodial epoxide during normal metabolic processes. (B) Cigarette smoke is a source of benzo[a]pyrene. (C) The activated (+)benzo[a]pyrene-7,8-dihydrodiol-9,10-epoxide can form a DNA adduct with guanine residues. (D) The benzo[a]pyrene adduct forms a bulky lesion that distorts the DNA backbone.
Figures from: (A) Eleska, (B) Elizabeth Aguilera (C) DI93biochemist, and (D) Zephryis
Back to the Top
DNA Crosslinking
Crosslinking of DNA occurs when various exogenous or endogenous agents react with two nucleotides of DNA, forming a covalent linkage between them. This crosslink can occur within the same strand (intrastrand) or between opposite strands of double-stranded DNA (interstrand) (Figure 12.9). These adducts interfere with cellular metabolism, such as DNA replication and transcription, triggering cell death.

Figure 12.9 Schematic Diagram of Intrastrand and Interstrand DNA Crosslinks.
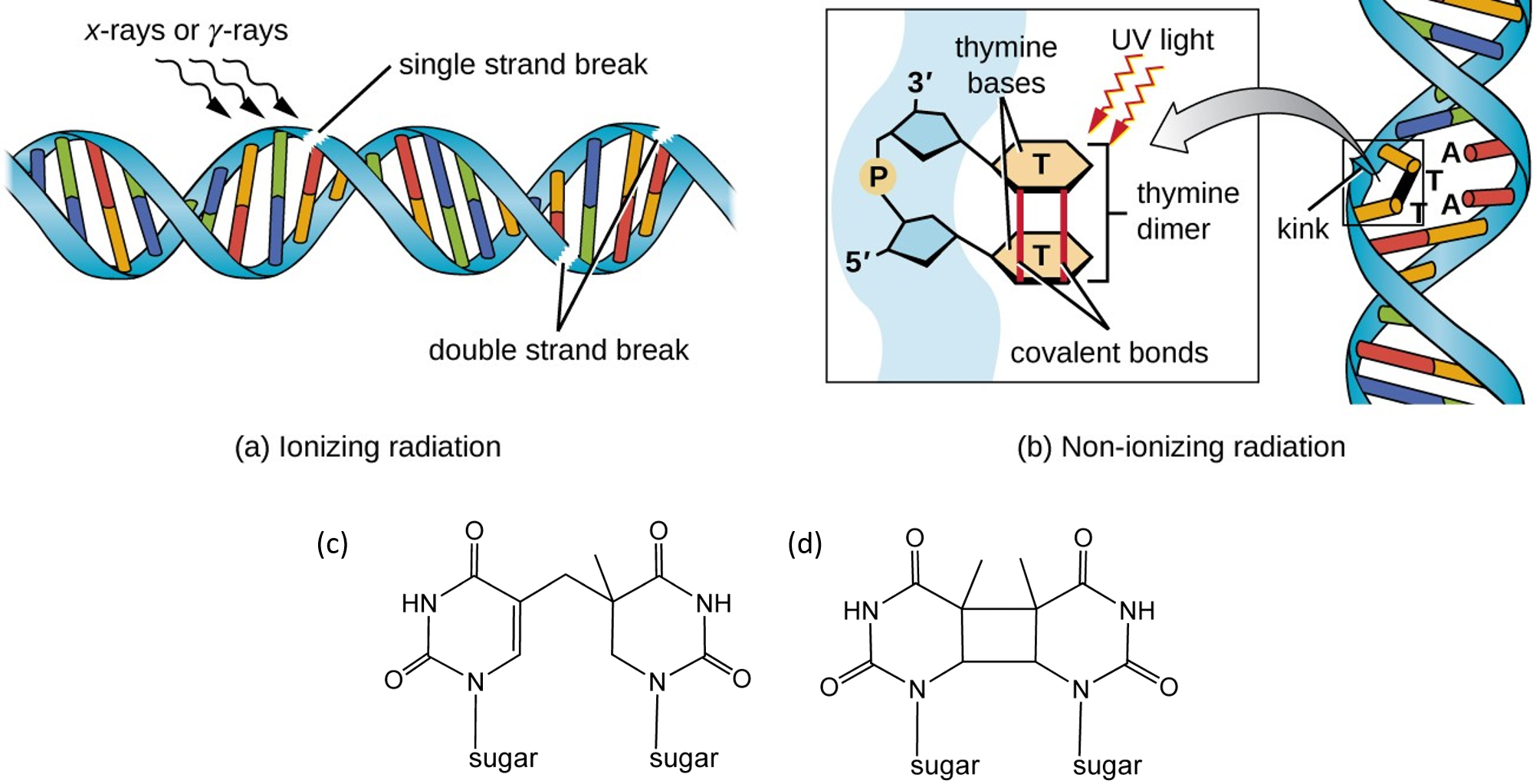
UV light can cause molecular crosslinks to form between two pyrimidine residues, commonly two thymine residues, that are positioned consecutively within a strand of DNA (Figure 12.10). Two common UV products are cyclobutane pyrimidine dimers (CPDs) and 6–4 photoproducts. These premutagenic lesions alter the structure and possibly the base-pairing. Up to 50–100 such reactions per second might occur in a skin cell during exposure to sunlight, but are usually corrected within seconds by photolyase reactivation or nucleotide excision repair. Uncorrected lesions can inhibit polymerases, cause misreading during transcription or replication, or lead to arrest of replication. Pyrimidine dimers are the primary cause of melanomas in humans.
{kind=link}
{kind=link}
{kind=link}
pyrene#/media/File:Benzo(a)pyrene_metabolism.svg){kind=link}
pyrene#/media/File:DG_rxn_with_BPDE.jpg){kind=link}
pyrene#/media/File:Benzopyrene_DNA_adduct_1JDG.png){kind=link}
Figure 12.10 Double-Stranded DNA breaks and Thymine Dimer Formation (a) Ionizing radiation, such as X-rays and γ-rays contain enough energy to cause single and double-stranded breaks in the DNA backbone. (b) Energy from non-ionizing radiation such as UV-light is directly absorbed by the DNA causing thymine residues that are adjacent within a single DNA strand to become crosslinked. (c) a 6,4-dimer that forms a single covalent bond and (d) thymine-thymine cyclobutane dimer that forms a two covalent bonds between the thymine residues.
Figures from: (a and b) Parker, et al (2019) Microbiology from Openstax and (c and d) Smokefoot
{kind=link}
{kind=link}
DNA Strand Breaks
Ionizing radiation such as that created by radioactive decay or in cosmic rays causes breaks in DNA strands (Figure 12.10a). Low-level ionizing radiation may induce irreparable DNA damage (leading to replicational and transcriptional errors needed for neoplasia or may trigger viral interactions) leading to premature aging and cancer. Chemical agents that form crosslinks within the DNA, especially interstrand crosslinks, can also lead to DNA strand breaks if the damaged DNA undergoes DNA replication. Crosslinked DNA can cause topoisomerase enzymes to stall in the transition state when the DNA backbone is in the cleaved state. Instead of relieving supercoiling and resealing the backbone, the stalled topoisomerase remains covalently linked to the DNA in a process called abortive catalysis. This leads to the formation of a single stranded break in the case of Top1 enzymes or double stranded breaks in the case of Top2 enzymes. DNA double strand breaks due to topoisomerase stalling can also occur during the transcription of DNA (Figure 12.11). In fact, abortive catalysis and the formation of DNA strand breaks during transcriptional events may serve as a damage sensor within the cell and help to instigate DNA damage response signaling pathways that initiate DNA repair processes.

Figure 12.11 Model of Topoisomerase IIB (TOP2B)-mediated DNA Double Strand Breaks During Transcription. (Top Diagram) in the uninduced state of transcription, Pol II is paused between +25 and +100 from the transcription start site. The pausing is attributed to different elements including pausing-stabilizing transcription factors, the +1 nucleosome, and DNA structure and torsion. Positive supercoiling ahead of Pol II may require the function of TOP2B. (Middle Diagram) Transcription activation induced by various stimuli activates TOP2B to resolve DNA torsion in the promoter and gene body. (Bottom Diagram) In this process, double strand breaks could be formed from abortive catalysis of TOP2B, which occurs frequently in some genes. This may be responsible for DNA damage response signaling that has been observed in a number of stimulus-inducible genes in humans.
Figure from: Morimoto, S., et al (2019) Genes 10:868
Back to the Top
12.3 Cellular Stress and DNA Damage Response
Genetic damage produced by either exogenous or endogenous mechanisms represents an ongoing threat to the cell. To preserve genome integrity, eukaryotic cells have evolved repair mechanisms specific for different types of DNA Damage. However, regardless of the type of damage a sophisticated surveillance mechanism, that elicits DNA damage checkpoints, detects and signals its presence to the DNA repair machinery. DNA damage checkpoints have been functionally conserved throughout eukaryotic evolution, with most of the relevant players in the checkpoint response highly conserved from yeast to humans. Checkpoints are induced to delay cell cycle progression and to allow cells time to repair damaged DNA prior to DNA replication (Figure 12.12). Once the damaged DNA is repaired, the checkpoint machinery triggers signals that will resume cell cycle progression. Within cells, multiple pathways contribute to DNA repair, but independent of the specific repair pathway involved, three phases of checkpoint activation are traditionally identified: (1) Sensing of damage, (2) Activating the signaling cascade, and (3) Switching on downstream effectors. The sensor phase recognizes the damage and activates the signal transduction phase to block cell cycle progression and select the appropriate repair pathway.
In multicellular organisms, the response to DNA damage can result in two major physiological consequences: (1) Cells can undergo cell cycle arrest, repair the damage and re-enter the cell cycle, or (2) cells can be targeted for cell death (apoptosis) and removed from the population. The cell cycle process is highly conserved and precisely controlled to govern the genome duplication and separation into the daughter cells. The cell cycle consists of four distinct and ordered phases, termed G0/G1 (gap 1), S (DNA synthesis), G2 (gap 2), and M (mitosis). Multiple checkpoints exist within each stage of the cell cycle to ensure the faithful replication of DNA in the S phase and the precise separation of the chromosomes into daughter cells. The G1 and G2 phases are critical regulatory checkpoints, whereby the restriction point between the G1 and S phase determines whether the cells enter the S phase or exit the cell cycle to halt at the G0 phase. The cell cycle progression requires the activity cyclin-dependent kinases (CDKs), a group of serine/threonine kinases. CDKs are activated when they form complexes with cyclin regulatory proteins that are expressed specifically at different stages of the cell cycle. Cyclins bind to and stabilize CDKs in their active conformation. The formation of cyclin/CDKs controls the cell-cycle progression via phosphorylation of the target genes, such as tumor suppressor protein retinoblastoma (Rb).
During DNA damage, the cell cycle is arrested or blocked by the action of cyclin-dependent kinase inhibitors. As noted in Figure 12.12, this is a complicated signal transduction cascade that has a number of downstream effects. A primary function of cell cycle arrest is that CDK inhibition allows time for DNA repair before cell-cycle progression into S-phase or mitosis. As shown in Figure 12.12, two major cell-cycle checkpoints respond to DNA damage; they occur pre- and post-DNA synthesis in the G1 and G2 phases, respectively, and impinge on the activity of specific CDK complexes. The checkpoint kinases phosphatidylinositol 3-kinase (PI3K)-like protein kinases (PI3KKs) ataxia telangiectasia and Rad3-related (ATR) or ataxia telangiectasia mutated (ATM) protein, and the transducer checkpoint kinases CHK1 (encoded by the CHEK1 gene) and CHK2 (encoded by the CHEK2 gene) are key regulators of DNA damage signaling. The DNA damage signaling is detected by ATM/ATR, which then phosphorylate and activate CHK2/CHK1, respectively. The activated CHK2 is involved in the activation of p53, leading to p53-dependent early phase G1 arrest to allow time for DNA repair. The activation of p53 induces the expression of the Cyclin-Dependent Kinase Inhibitor (CKI) p21CIP1 gene, leading to inhibition of cyclin E/CDK2 complexes and subsequent upregulation of DNA repair machinery.
If the DNA repair cannot be completed successfully or the cells cannot program to respond to the stresses of viable cell-cycle arrest, the cells face the fate of apoptosis induced by p53. The activated CHK1 mediates temporary S phase arrest through phosphorylation to inactivate CDC25A, causing ubiquitination and proteolysis. Moreover, the activated CHK1 phosphorylates and inactivates CDC25C, leading to cell-cycle arrest in the G2 phase. The active CHK1 also directly stimulates the phosphorylation of WEE1, resulting in enhancing the inhibitory Tyr15 phosphorylation of CDK2 and CDK1 and subsequent cell-cycle blocking in G2 phase. The activity of WEE1 can also be stimulated by the low levels of CDK activity in G2 cell-cycle phase. The SAC, also known as the mitotic checkpoint, functions as the monitor of the correct attachment of the chromosomes to the mitotic spindle in metaphase, which is regulated by the TTK protein kinase (TTK, also known as monopolar spindle 1 (MPS1)). The activation of SAC transiently induces cell-cycle arrest via inhibiting the activation of APC/C. In order to establish and maintain the mitotic checkpoint, the TTK recruits many checkpoint proteins to kinetochores during mitosis via phosphorylating its substrates to ensure adequate chromosome segregation and genomic integrity. In this way, the genomic instability from chromosome segregation defects is protected by SAC. Once the SAC is passed, the APC/C E3 ligase complex stimulates and tags cyclin B and securin for ubiquitin-mediated degradation, leading to the initiation of mitosis. In a word, the checkpoints offer a failsafe mechanism to ensure the genomic integrity from the parental cell to daughter cell. The signal transduction cascade of checkpoint activation eventually converges to CDK inhibition, which indicates the CDK function as a key driver of cell-cycle progression.
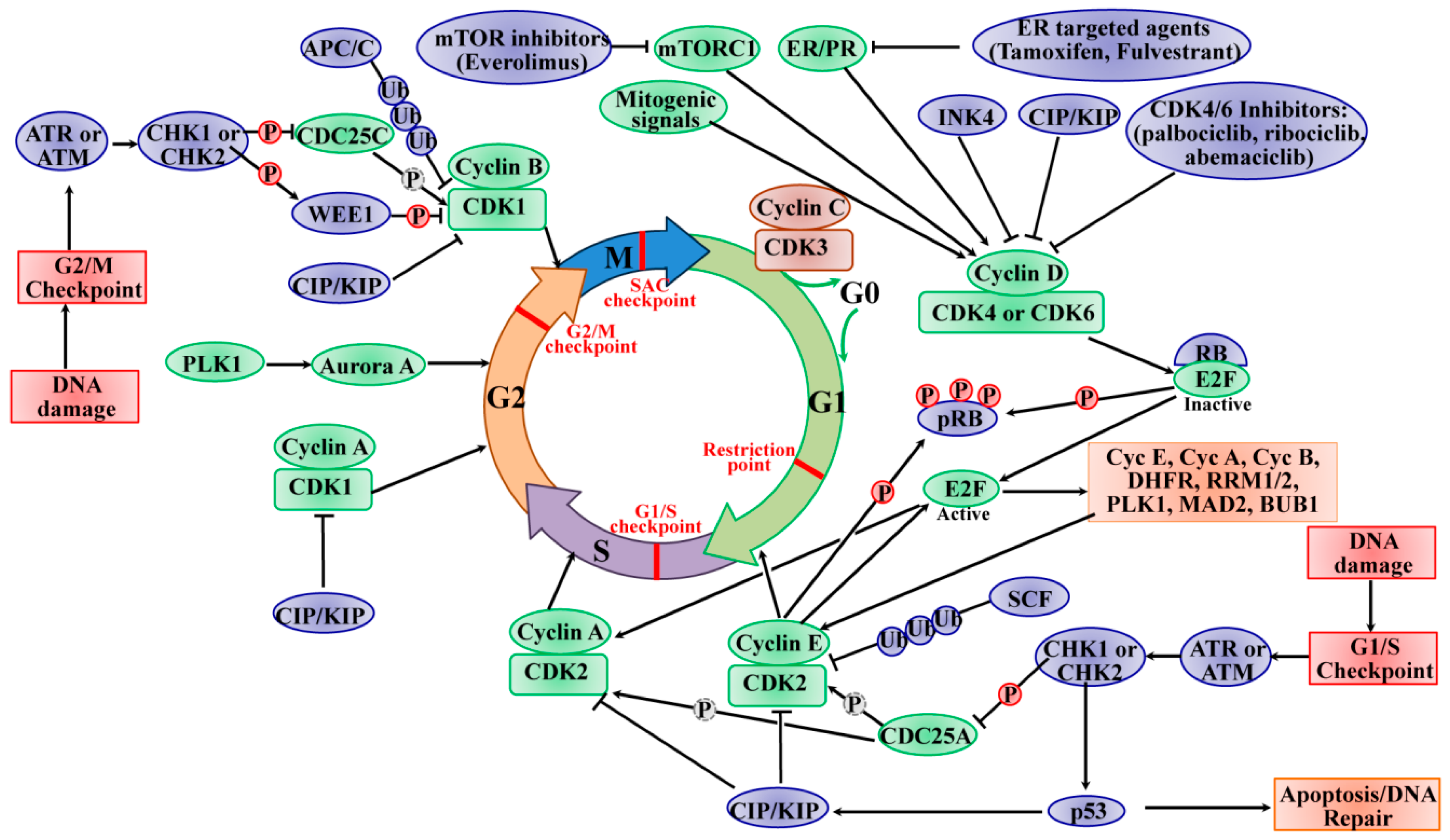
Figure 12.12 Cell Fates Following DNA Damage. Cell cycle checkpoints are induced by DNA damage, shown in red. Cyclin-Dependent Kinase Inhibitors (CIP/KIP), shown in purple, block cell cycle progession in all phases of the cell cycle (G1, S, G2, or M) following DNA damage by inhibiting cyclin-dependent kinase complexes (shown in green). Signaling cascades activated in response to DNA damage also elicit DNA repair pathways, or if the DNA damage is severe, programmed cell death (Apoptosis) will be activated.
Image from: Ding, L., et al. (2020) Int. J. Mol. Sci. 21(6):1960
Back to the Top
In addition to blocking cell cycle progression, DNA damage sensors also activate DNA repair mechanisms that are specific for the type of damage present. For example, single stranded DNA breaks are repaired primarily by Base Excision Repair, bulky DNA adducts and crosslinks are repaired by Nucleotide Excision Repair, and smaller nucleotide mutations, such as alkylation are repaired by Mismatch Repair. Cells also have two major mechanisms for repairing Double-Strand-Breaks (DSBs). They include Non-Homologous End-Joining (NHEJ) and Homologous Recombination (HR). If damage is too extensive to be repaired, apoptotic pathways will be elicited. In the following sections, details about the major DNA repair pathways will be given.
12.4 Mismatch Repair
DNA mismatch repair (MMR) is a highly conserved DNA repair system (Table 12.2) that greatly contributes to maintain genome stability through the correction of mismatched base pairs and small modifications of DNA bases, such as alkylation. The major source of mismatched base pairs is replication error, although it can arise also from other biological processes. Thus, the MMR machinery must have a mechanism for determining which strand of the DNA is the template strand and which strand has been newly synthesized. In E. coli, methylation of the DNA is a common post-replicative modification that occurs. Thus, in newly synthesized DNA, the unmethylated strand is recognized as the new strand and the methylated strand is used as the template to repair mismatches. In E.coli, MMR increases the accuracy of DNA replication by 20–400-fold. Mutations and epigenetic silencing in MMR genes have been implicated in up to 90% of human hereditary nonpolyposis colon cancers, indicating the significance of this repair system in maintaining genomic stability. Post-replicative MMR is performed by the long-patch MMR mechanism in which multiple proteins are involved and a relatively long tract of the oligonucleotide is excised during the repair reaction. In contrast, particular kinds of mismatched base pairs are repaired through very short-patch MMR in which a short oligonucleotide tract is excised to remove the lesion.
Table 12.2 Mismatch Repair Enzymes in Bacteria, Yeast, and Humans
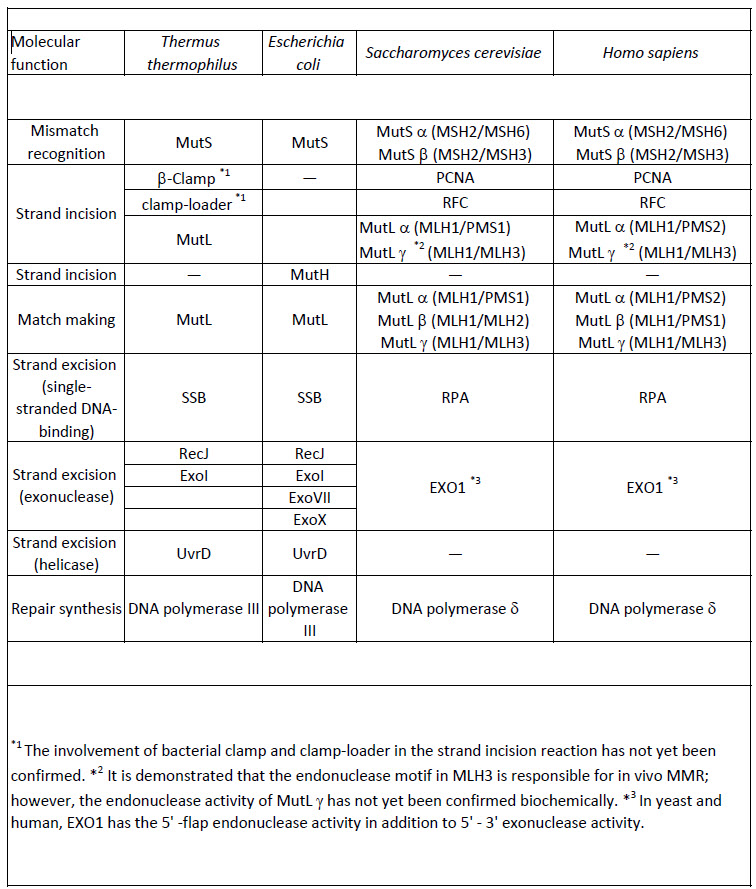
Currently, two types of MMR mechanisms have been elucidated: one is expected to be employed by eukaryotes and the majority of bacteria, and the other is specific to E. coli and closely related bacteria. As shown in Figures 12.13(a) and 12.13(b), MMR in eukaryotes and most bacteria directs the repair to the error-containing strand of the mismatched duplex by recognizing the strand discontinuities. On the other hand, as shown in Figure 12.13(c), E. coli MMR reads the absence of methylation as a strand discrimination signal. In both MMR systems, strand discrimination is conducted by nicking endonucleases. MutL homologues from eukaryotes and most bacteria incise the discontinuous strand to introduce the entry or termination point for the excision reaction. In E. coli, MutH nicks the unmethylated strand of the duplex to generate the entry point of excision. Following the excision of the damaged or mismatched strand, helicases (such as UvrD) and exonucleases (such as ExoI) excise a short region of nucleotides from the damaged strand. DNA pol III or DNA pol δ fills in the gap and DNA ligase repairs the backbone.

Figure 12.13 A schematic representation of MMR pathway models. (a) Eukaryotic MMR. A DNA mismatch is generated by the misincorporation of a base during DNA replication. MutSα recognizes base-base mismatches. MutLα nicks the 3′- or 5′-side of the mismatched base on the discontinuous strand. The resulting DNA segment is excised by the EXO1 exonuclease, in cooperation with the single-stranded DNA-binding protein RPA. The DNA strand is resynthesized by DNA polymerase δ and DNA ligase. (b) MMR in mutH-less bacteria. Mismatched bases are recognized by MutS. After the incision of discontinuous strand by MutL, the error-containing DNA strand is removed by the cooperative functions of DNA helicases, such as UvrD, the exonucleases RecJ and ExoI, and the single-stranded DNA-binding protein SSB. DNA polymerase III and DNA ligase fill the gap to complete the repair. (c) E. coli MMR. MutS recognizes mismatched bases, and MutL interacts with and stabilizes the complex. Then, MutH endonuclease is activated to incise the unmethylated GATC site to create an entry point for the excision reaction. DNA helicase, a single-stranded DNA-binding protein, and several exonucleases are involved in the excision reaction.
Figure from: Fukui, K. (2010) J. Nuc. Acids 260512
Back to the Top
12.5 Base Excision Repair
Most oxidized bases are removed from DNA by enzymes operating within the Base Excision Repair (BER) pathway. Single stranded DNA breaks can also be repaired through this process. Removal of oxidized bases in DNA is fairly rapid. For example, 8-oxo-dG was increased 10-fold in the livers of mice subjected to ionizing radiation, but the excess 8-oxo-dG was removed with a half-life of 11 minutes. 8-oxoG is excised by 8-oxoguanine DNA glycosylase (OGG1) leaving an apurinic site (AP site) (Figure 12.14). AP sites are then processed further into single strand breaks via backbone incision of AP-endonuclease 1 (APE1). In long patch base excision repair, the base and some additional nucleotides are replaced dependent on the activity of polymerase delta (Polδ) and epsilon (Polε) together with proliferating cell nuclear antigen (PCNA). The old strand is removed by Flap-endonuclease 1 (FEN1), before ligase I (LigI) ligates the backbone back together. Short patch base excision repair constitutes of polymerase beta (Polβ) replacing the single missing base, ligase III (LigIII) ligating the DNA backbone back together, and X-ray repair cross-complementing protein 1 (XRCC1) aiding the process and serving as a scaffold for additional factors.

Figure 12.14 Base Excision Repair. Base excision repair (BER) of 8-oxo-7,8-dihydroguanine (8-oxoG). Oxidative DNA damage is repaired via several repair intermediates by base excision repair (BER). Through removal of the oxidized base, a reactive apurinic site (AP site) is formed. Incision of the strand creates a single strand break, and the damaged site is then repaired through either short or long patch BER
Figure from: Poetsch, A.R. (2020) Comp & Struct Biotech J. 18:207-219.
12.6 Nucleotide Excision Repair
Bulky DNA adducts and DNA crosslinks, such as those caused by UV light are repaired using Nucleotide Excision Repair (NER) pathways. In higher eukaryotic cells, NER excises 24-32 nucleotide DNA fragments containing the damaged lesion with extreme accuracy. Reparative synthesis using the undamaged strand as a template, followed by ligation of the single strand break that emerged as a result of the damage, is the final stage of DNA repair. The process involves the coordinated action of approximately 30 proteins that successively form complexes with variable compositions on the DNA. NER consists of two pathways distinct in terms of initial damage recognition. Global genome nucleotide excision repair (GG-NER) detects and eliminates bulky damages in the entire genome, including the untranscribed regions and silent chromatin, while transcription-coupled nucleotide excision repair (TC -NER) operates when damage to a transcribed DNA strand limits transcription activity. TC-NER is activated by the stalling of RNA polymerase II at the damaged sites of a transcribed strand, while GG-NER is controlled by the protein, XPC, a specialized protein factor that reveals the damage. A schematic GG-NER process is presented in Figure 12.15.

Figure 12.15 Schematic of Global Genome Nucleotide Excision Repair.
Figure from: Petruseva, I.O., et al (2014) 6(1):23-34.
Genetic mutations in NER pathway genes can result in UV-sensitive and high-carcinogenic pathologies, such as xeroderma pigmentosum (XP), the Cockayne syndrome (CS), and trichothiodystrophy (TTD), as well as some neurodegenerative manifestations.
Xeroderma pigmentosum has provided the names of some of the genes involved in NER. Mutation of XP genes and loss of proper NER function cause the symptoms associated with the disease. People with XP have an impaired ability to repair bulky DNA adducts and crosslinks, such as thymine dimers that are caused by UV-light exposure. People suffering from XP have extreme photosensitivity, skin atrophy, hyperpigmentation and a high rate of sunlight-induced skin cancer (Figure 12.16). The risk of internal tumors in XP patients is also 1,000-fold higher. Moreover, the disease is often associated with neurologic disorders. Currently, there is no effective treatment for this disorder.
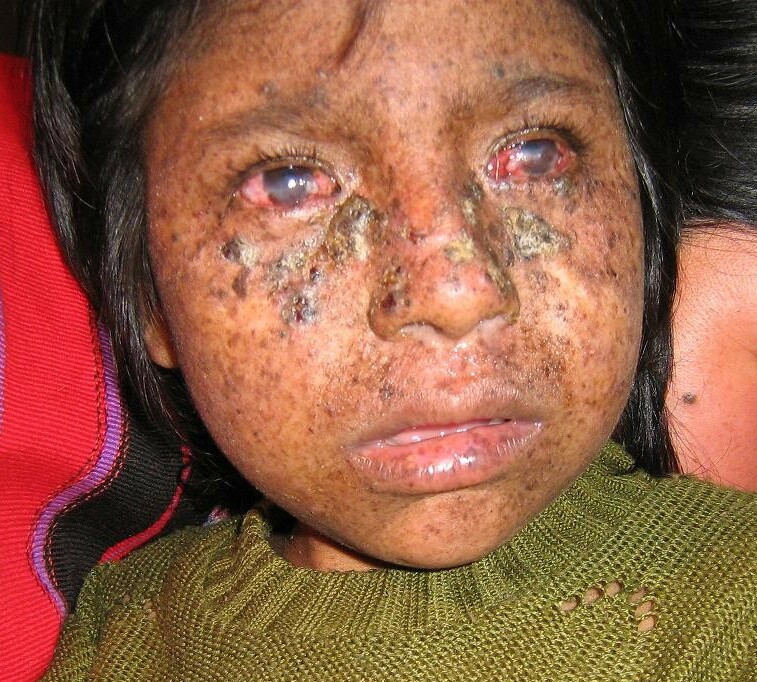
Figure 12.16 An eight year old girl from Guatemala with Xeroderma Pigmentosum. Frontal image of face, showing large hyperkeratotic lesions with some induration suspicious of actinic keratosis and early squamous cell carcinoma. Also numerous hyper-pigmented lentigos and xerosis on the entire front of the face. Marked corneal scarring and injection is evident.
{kind=link}
The detection of bulky DNA lesions during NER is particularly challenging for a cell, which can be solved only through highly sensitive recognition that require multiple protein components. In contrast to BER, where a damaged base is simultaneously recognized and eliminated by a single specialized glycosylase, specialized groups of proteins are responsible for recognition of the lesion and the excision of the lesion in NER. In eukaryotic NER, universal sensor proteins perform the initial recognition of the total range of bulky damages. In the case of TC-NER, it occurs when the transcribing RNA polymerase II is stalled by damage; in GG-NER, these are complexes of the XPC factor and DDB1-DDB2 heterodimer (XPE factor) enhancing the repair of UV damage (Fig 12.15). In general, NER recognition of damage is a multistep process involving several proteins that form near damaged complexes of variable compositions. The process is completed by the formation of a preincision complex ready to eliminate a damaged DNA fragment by specialized NER endonucleases.
In a eukaryotic cell after stable XPC/DNA complex formation during the initial recognition of the damage, NER is actually performed by a repairasome, which is a complex of variable composition and architecture consisting of a large number of subunits. Individual subunits of the complex have no sufficient affinity and selectivity to the substrate (DNA containing bulky damage). The situation changes when specific protein complexes are established at the damage site. The NER proteins of these complexes are joined by the DNA processing. A total of 18 polypeptides must be accurately positioned within two or three DNA turns when a stable structure ready for damage removal is formed and excision starts. The structure of NER-associated proteins provides the possibility of contact with the DNA substrate and of dynamic specific protein-protein interactions. The changes in interactions performed by the same protein are one of the mechanisms that regulate the repair process and fine-tune the complexes, providing high-precision nucleotide excision repair.
Back to the Top
12.7 Repair of Double-Stranded DNA Breaks
Cells have evolved two main pathways to repair double-strand breaks within the DNA: the non-homologous end-joining (NHEJ) pathway, that ensures direct resealing of DNA ends; and the homologous recombination (HR) pathway that relies on the presence of homologous DNA sequences for DSB repair (Figure 2.16)
NHEJ repair is the simplest and most widely utilized mechanism to repair DSB that occur in DNA. Repair by NHEJ involves direct resealing of the two broken ends independently of sequence homology. Although being active throughout the cell cycle, NHEJ is relatively more important during G1 phase. Proteins required for NHEJ include, but are not restricted to, the highly conserved Ku70/Ku80 heterodimeric complex, DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and DNA Ligase IV (LIG4) in complex with XRCC4. By directly binding DNA ends, Ku70/Ku80 ensures protection against exonucleases and, as such, acts as an inhibitor of HR. Very short sequence homologies are likely to help DNA end alignment prior to NHEJ-dependent repair, however, they are not strictly required. NHEJ protects genetic integrity by rejoining broken strands of DNA that may otherwise be lost during DNA replication and cell regeneration. However, during the process of NHEJ, insertions or deletions within the joined regions may occur (Fig 2.16).

Figure 2.16 Main Pathways of DNA Double-Stranded Break Repair. Non-homologous end-joining (NHEJ) and homologous recombination (HR) pathways act competitively to repair DNA double-strand breaks (DSBs). Key players of NHEJ and HR are depicted. The MRE11/RAD50/XRS2 (MRX) complex is recruited very early at DNA ends and appears to play important roles for both NHEJ and HR. Ku70/Ku80 heterodimer is required for NHEJ and, through inhibition of DNA end resection (5′–3′ exo), acts as a repressor of HR. Fidelity of NHEJ-dependent DSB repair is low and, most of the time, associated with nucleotide deletions and/or insertions at repair junctions. The common early step of HR-dependent mechanisms is the formation of ssDNA which is then coated by replication protein A (RPA). Single-strand annealing (SSA) mechanism requires the presence of direct repeats (shown in orange) on both sides of the break. SSA does not imply any strand invasion process and is therefore not dependent on RAD51 protein. Strand invasion and D-loop formation are however common steps of synthesis-dependent strand annealing (SDSA) and double Holliday junction (HJ) dissolution mechanisms. In the latter case, double Holliday junctions are resolved with or without crossing-over.
Figure from: Diccotignies, A. (2013) Frontiers in Genetics 4(48):48
In contrast to NHEJ, homologous recombination (HR) requires a homologous DNA sequence to serve as a template for DNA-synthesis-dependent repair and involves extensive DNA-end processing. As expected, HR is extremely accurate, as it leads to precise repair of the damaged locus using DNA sequences homologous to the broken ends. HR predominantly uses the sister chromatid as a template for DSB repair, rather than the homologous chromosome. Correspondingly, HR is largely inhibited while cells are in the G1 phase of the cell cycle, when the sister chromatid has not yet been replicated (Fig 2.17A). HR repair mechanisms play a bigger role in DSB repair that occurs after S-phase DNA replication (S-phase, G2, and M).
Repair through HR is not defined by a unique mechanism but operates through various mechanistically distinct DSB repair processes, including synthesis-dependent strand annealing (SDSA), double Holliday junction resolution, and single-strand annealing (SSA). The common step for HR-dependent DSB repair mechanisms is the initial formation of single-stranded DNA (ssDNA) for pairing with homologous DNA template sequences. For this to occur, the 5′ DNA strand at the DSB is processed by multiple nucleases and accessory proteins to create a 3′ ssDNA section that can be used as a template for recombination (Fig. 2.16).
Figure 2.17B provides a more detailed look at the HR process. During the highly regulated process of HR, three main phases can be distinguished. Firstly, 3′-single-stranded DNA (ssDNA) ends are generated by nucleolytic degradation of the 5′-strands. This first step is catalyzed by endonucleases, including the MRN complex (consisting of Mre11, Rad50, and Nbs1). In a second step, the ssDNA-ends are coated by replication protein A (RPA) filaments. In a third step, RPA is replaced by Rad51 in a BRCA1- and BRCA2-dependent process, to ultimately perform the recombinase reaction using a homologous DNA template.
Importantly, HR is not only employed to repair DNA lesions induced by DNA damaging agents, but is also essential for proper chromosome segregation during meiosis. The relevance of HR in these physiological processes is illustrated by its strict requirement during development. Mice lacking key HR genes, such as Brca1, Brca2, or Rad51, display extensive genetic alterations which lead to early embryonic lethality. Whereas homozygous inactivation of HR genes is usually embryonic lethal, heterozygous inactivation of for instance, BRCA1 and BRCA2, does not interfere with cellular viability but rather predisposes individuals to cancer, including breast and ovarian cancer. The tumors that develop in individuals with heterozygous BRCA1/2 mutations invariably lose their second BRCA1/2 allele, indicating that in certain cancers, the absence of BRCA1/2 is compatible with cellular proliferation. How exactly such tumors cope with their HR defect is currently not fully understood.
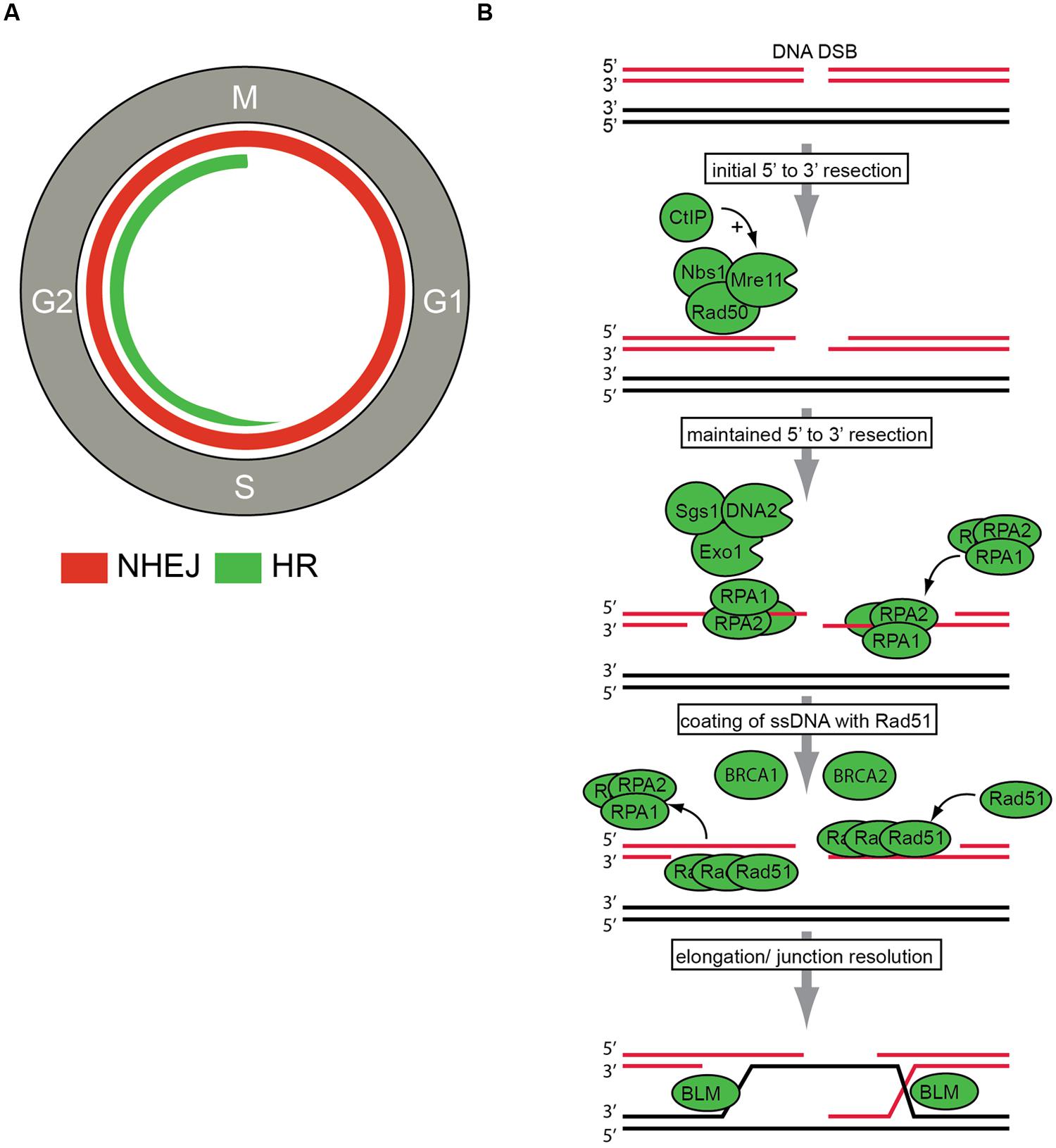
Figure 2.17 DNA double strand break (DSBs) repair. (A) DNA DSBs repair pathways in the context of cell cycle regulation. Non-homologous end joining (NHEJ) can be performed throughout the cell cycle and is indicated with the red line. Homologous recombination (HR) can only be employed in S/G2 phases of the cell cycle and is indicated in green. (B) The key steps in HR repair pathway are indicated. After DSB recognition, 5′–3′ end resection is initiated by the MRN (Mre11, Rad50, Nbs1) complex and CtIP. Subsequently, further resection by the Exo1, DNA2, and Sgs1 proteins is conducted to ensure ‘maintained’ resection. Then, resected DNA-ends are bound by replication protein A (RPA). The actual recombination step within HR repair, termed strand exchange, is executed by the recombinase Rad51. Rad51 replaces RPA to eventually assemble helical nucleoprotein filaments called ‘presynaptic filaments.’ This process is facilitated by other HR components, including BRCA1 and BRCA2. Final step of junction resolution is executed by helicases including Bloom syndrome, RecQ helicase-like (BLM) helicase.
Figure from: Krajewska, M., Fehrmann, R.S.N., de Vries, E.G.E., and van Vugt, A.A.T.M. (2015) Front. Genet. 6:96
Back to the Top
12.8 Error-Prone Bypass and Translesion Synthesis
If DNA is not repaired prior to DNA replication, the cell must employ another strategy to replicate the DNA, even in the presence of a DNA lesion. This is important to avoid causing double stranded DNA breaks that can occur when a replisome stalls at the replication fork. Under these circumstances, another strategy that cells use to respond to DNA damage is to bypass lesions found during DNA replication and continue with the replicative process. DNA damage bypass can occur by recombination mechanisms or through a novel mechanism called translesion synthesis. Translesion synthesis employs an alternate DNA polymerase that can substitute for a DNA polymerase that has stalled at the replication fork due to DNA damage. Specialized DNA polymerases, that are active in regions with DNA damage, have active sites that can accommodate fluctuations in DNA topography that enable them to bypass the lesions and continue with the replicative process.
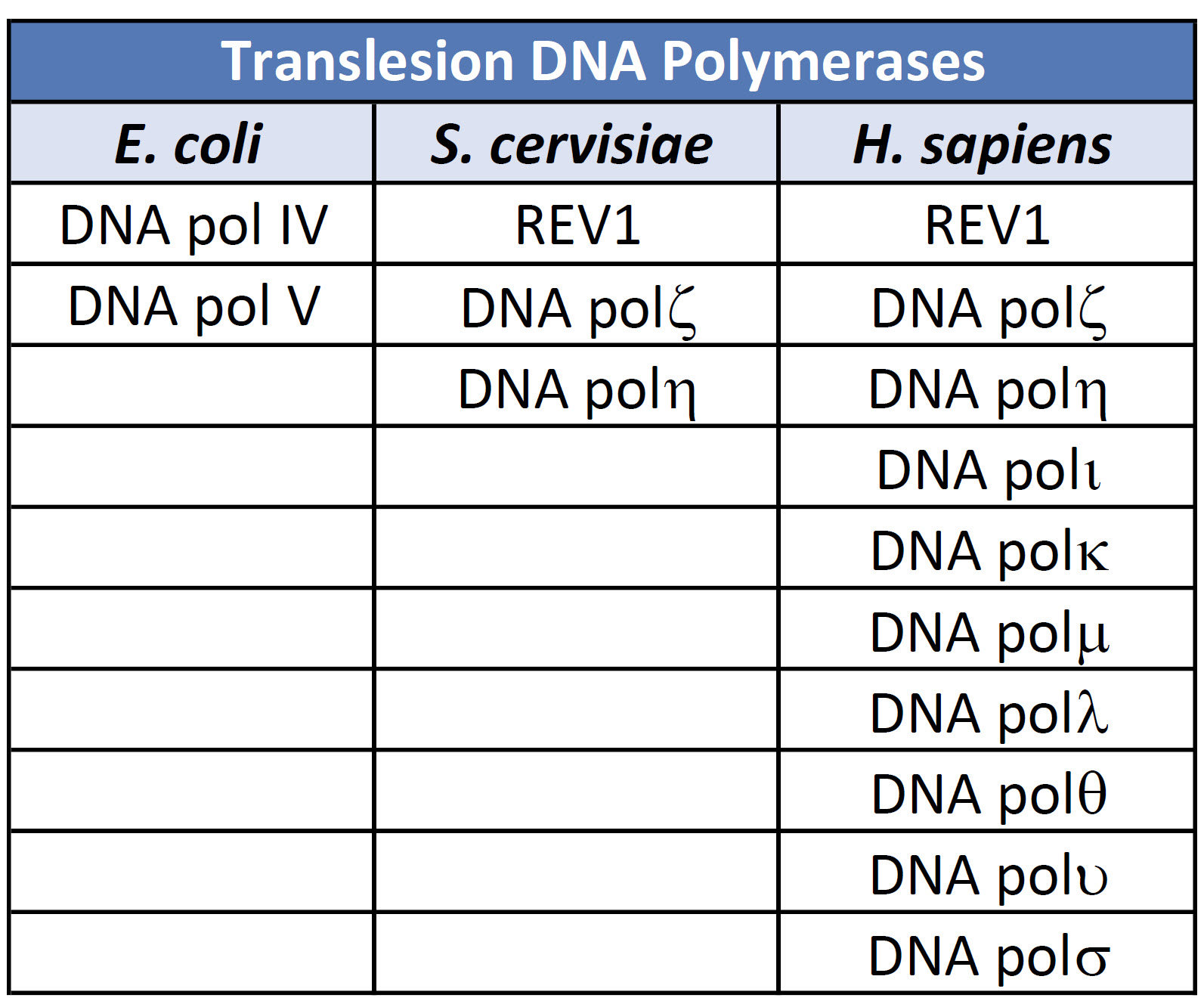
The evolution of DNA polymerases that can tolerate the presence of distorted DNA lesions and continue with the replicative process can be seen at all levels of life, from prokaryotic, single-celled organisms all the way through eukaryotic multicellular organisms, including humans. In fact, within vertebrates, there has been a large expansion of DNA polymerases that play a role in DNA damage bypass mechanisms and highlight the importance of these processes in damage tolerance and cell survival (Table 12.3).
Table 12.3 DNA Polymerases involved in Error-Prone Bypass
The activity of error prone DNA polymerases is tightly regulated to avoid the rampant introduction of mutations within the DNA sequence. One of the main mechanisms that is employed within a replisome that is stalled at the replication fork due to DNA damage, involves the monoubiquitination of PCNA. Recall from Chapter 9, that PCNA is the sliding clamp that enables the DNA polymerase to bind tightly enough with the DNA during replication to mediate efficient DNA synthesis. Monoubiquitination of PCNA enables the recruitment of a translesion DNA polymerases and the bypass of the damaged lesions during DNA synthesis.
During translesion synthesis, the polymerase must insert a dNTP opposite of the lesion. It is likely that none of the dNTP bases will be able to form stable hydrogen bond interactions with the damaged lesion. Thus, the nucleotide that causes the least distortion or repulsion will usually be added across from the lesion. This can cause transition or transversion mutations to occur at the lesion location. Alternatively, translesion polymerases can be prone to slippage, and either cause an insertion or deletion mutation in the vicinity of the DNA lesion. These slippages can lead to frameshift mutations if they occur within gene coding regions. Thus, over a lifetime, translesion synthesis in multicellular organisms can lead to an accumulation of mutations within somatic cells and cause the formation of tumors and the disease of cancer.
Evolution by natural selection is also possible due to random mutations that occur within germ cells. Occasionally, germline mutations may lead to a beneficial mutation that enhances the survival of an individual within a population. If this gene proves to enhance the survival of the population, it will be selected for over time within the population and cause the evolution of that species. An example of a beneficial mutation is the case a population of people that show resistance to HIV infection. Since the first case of infection with human immunodeficiency virus (HIV) was reported in 1981, nearly 40 million people have died from HIV infection, the virus that causes acquired immune deficiency syndrome (AIDS). The virus targets helper T cells that play a key role in bridging the innate and adaptive immune response, infecting and killing cells normally involved in the body’s response to infection. There is no cure for HIV infection, but many drugs have been developed to slow or block the progression of the virus. Although individuals around the world may be infected, the highest prevalence among people 15–49 years old is in sub-Saharan Africa, where nearly one person in 20 is infected, accounting for greater than 70% of the infections worldwide (Figure 2.18). Unfortunately, this is also a part of the world where prevention strategies and drugs to treat the infection are the most lacking.

Figure 2.18 HIV is highly prevalent in sub-Saharan Africa, but its prevalence is quite low in some other parts of the world.
Figure from: Parker, et al (2019) Microbiology from Openstax
In recent years, scientific interest has been piqued by the discovery of a few individuals from northern Europe who are resistant to HIV infection. In 1998, American geneticist Stephen J. O’Brien at the National Institutes of Health (NIH) and colleagues published the results of their genetic analysis of more than 4,000 individuals. These indicated that many individuals of Eurasian descent (up to 14% in some ethnic groups) have a deletion mutation, called CCR5-delta 32, in the gene encoding CCR5. CCR5 is a coreceptor found on the surface of T-cells that is necessary for many strains of the virus to enter the host cell. The mutation leads to the production of a receptor to which HIV cannot effectively bind and thus blocks viral entry. People homozygous for this mutation have greatly reduced susceptibility to HIV infection, and those who are heterozygous have some protection from infection as well.
It is not clear why people of northern European descent, specifically, carry this mutation, but its prevalence seems to be highest in northern Europe and steadily decreases in populations as one moves south. Research indicates that the mutation has been present since before HIV appeared and may have been selected for in European populations as a result of exposure to the plague or smallpox. This mutation may protect individuals from plague (caused by the bacterium Yersinia pestis) and smallpox (caused by the variola virus) because this receptor may also be involved in these diseases. The age of this mutation is a matter of debate, but estimates suggest it appeared between 1875 years to 225 years ago, and may have been spread from Northern Europe through Viking invasions.
This exciting finding has led to new avenues in HIV research, including looking for drugs to block CCR5 binding to HIV in individuals who lack the mutation. Although DNA testing to determine which individuals carry the CCR5-delta 32 mutation is possible, there are documented cases of individuals homozygous for the mutation contracting HIV. For this reason, DNA testing for the mutation is not widely recommended by public health officials so as not to encourage risky behavior in those who carry the mutation. Nevertheless, inhibiting the binding of HIV to CCR5 continues to be a valid strategy for the development of drug therapies for those infected with HIV.
Back to the Top
12.9 Practice Problems
Multiple Choice
Which of the following is a change in the sequence that leads to formation of a stop codon?
- missense mutation
- nonsense mutation
- silent mutation
- deletion mutation
[reveal-answer q=”745512″]Show Answer[/reveal-answer]
[hidden-answer a=”745512″]Answer b. A nonsense mutation is a change in the sequence that leads to formation of a stop codon.[/hidden-answer]
The formation of pyrimidine dimers results from which of the following?
- spontaneous errors by DNA polymerase
- exposure to gamma radiation
- exposure to ultraviolet radiation
- exposure to intercalating agents
[reveal-answer q=”709151″]Show Answer[/reveal-answer]
[hidden-answer a=”709151″]Answer c. The formation of pyrimidine dimers results from exposure to ultraviolet radiation.[/hidden-answer]
Which of the following is an example of a frameshift mutation?
- a deletion of a codon
- missense mutation
- silent mutation
- deletion of one nucleotide
[reveal-answer q=”688366″]Show Answer[/reveal-answer]
[hidden-answer a=”688366″]Answer a. The deletion of one nucleotide is an example of a frameshift mutation.[/hidden-answer]
Which of the following is the type of DNA repair in which thymine dimers are directly broken down by the enzyme photolyase?
- direct repair
- nucleotide excision repair
- mismatch repair
- proofreading
[reveal-answer q=”755583″]Show Answer[/reveal-answer]
[hidden-answer a=”755583″]Answer a. In a direct repair, thymine dimers are directly broken down by the enzyme photolyase.[/hidden-answer]
Which of the following regarding the Ames test is true?
- It is used to identify newly formed auxotrophic mutants.
- It is used to identify mutants with restored biosynthetic activity.
- It is used to identify spontaneous mutants.
- It is used to identify mutants lacking photoreactivation activity.
[reveal-answer q=”770537″]Show Answer[/reveal-answer]
[hidden-answer a=”770537″]Answer b. It is used to identify mutants with restored biosynthetic activity.[/hidden-answer]
Fill in the Blank
A chemical mutagen that is structurally similar to a nucleotide but has different base-pairing rules is called a ________.
[reveal-answer q=”702924″]Show Answer[/reveal-answer]
[hidden-answer a=”702924″]A chemical mutagen that is structurally similar to a nucleotide but has different base-pairing rules is called a nucleoside analog.[/hidden-answer]
The enzyme used in light repair to split thymine dimers is called ________.
[reveal-answer q=”939657″]Show Answer[/reveal-answer]
[hidden-answer a=”939657″]The enzyme used in light repair to split thymine dimers is called photolyase.[/hidden-answer]
The phenotype of an organism that is most commonly observed in nature is called the ________.
[reveal-answer q=”640686″]Show Answer[/reveal-answer]
[hidden-answer a=”640686″]The phenotype of an organism that is most commonly observed in nature is called the wild type.[/hidden-answer]
True/False
Carcinogens are typically mutagenic.
[reveal-answer q=”166576″]Show Answer[/reveal-answer]
[hidden-answer a=”166576″]True[/hidden-answer]
Think about It
Why is it more likely that insertions or deletions will be more detrimental to a cell than point mutations?
Critical Thinking
Below are several DNA sequences that are mutated compared with the wild-type sequence: 3′-T A C T G A C T G A C G A T C-5′. Envision that each is a section of a DNA molecule that has separated in preparation for transcription, so you are only seeing the template strand. Construct the complementary DNA sequences (indicating 5′ and 3′ ends) for each mutated DNA sequence, then transcribe (indicating 5′ and 3′ ends) the template strands, and translate the mRNA molecules using the genetic code, recording the resulting amino acid sequence (indicating the N and C termini). What type of mutation is each?
| Mutated DNA Template Strand #1: 3′-T A C T G T C T G A C G A T C-5′ | |
|---|---|
| Complementary DNA sequence: | [practice-area rows=”1″][/practice-area] |
| mRNA sequence transcribed from template: | [practice-area rows=”1″][/practice-area] |
| Amino acid sequence of peptide: | [practice-area rows=”1″][/practice-area] |
| Type of mutation: | [practice-area rows=”1″][/practice-area] |
| Mutated DNA Template Strand #2: 3′-T A C G G A C T G A C G A T C-5′ | |
|---|---|
| Complementary DNA sequence: | [practice-area rows=”1″][/practice-area] |
| mRNA sequence transcribed from template: | [practice-area rows=”1″][/practice-area] |
| Amino acid sequence of peptide: | [practice-area rows=”1″][/practice-area] |
| Type of mutation: | [practice-area rows=”1″][/practice-area] |
| Mutated DNA Template Strand #3: 3′-T A C T G A C T G A C T A T C-5′ | |
|---|---|
| Complementary DNA sequence: | [practice-area rows=”1″][/practice-area] |
| mRNA sequence transcribed from template: | [practice-area rows=”1″][/practice-area] |
| Amino acid sequence of peptide: | [practice-area rows=”1″][/practice-area] |
| Type of mutation: | [practice-area rows=”1″][/practice-area] |
| Mutated DNA Template Strand #4: 3′-T A C G A C T G A C T A T C-5′ | |
|---|---|
| Complementary DNA sequence: | [practice-area rows=”1″][/practice-area] |
| mRNA sequence transcribed from template: | [practice-area rows=”1″][/practice-area] |
| Amino acid sequence of peptide: | [practice-area rows=”1″][/practice-area] |
| Type of mutation: | [practice-area rows=”1″][/practice-area] |
12.9 References
- World Health Organization. ” Global Health Observatory (GHO) Data, HIV/AIDS.“ http://www.who.int/gho/hiv/en/. Accessed August 5, 2016. ↵
- Parker, N., Schneegurt, M., Thi Tu, A-H., Lister, P., Forster, B.M. (2019) Microbiology. Openstax. Available at: https://opentextbc.ca/microbiologyopenstax/
- Wikipedia contributors. (2020, July 15). DNA oxidation. In Wikipedia, The Free Encyclopedia. Retrieved 03:44, July 16, 2020, from https://en.wikipedia.org/w/index.php?title=DNA_oxidation&oldid=967811859
- Wakim, S. and Grewal, M. (2020) Human Biology. Libretexts. Available at: https://bio.libretexts.org/Bookshelves/Human_Biology/Book%3A_Human_Biology_(Wakim_and_Grewal)
- Ahmad, A., Nay, S.L. and O’Conner, T.R. (2015) Chapter 4 Direct Reversal Repair in Mammalian Cells. Published through INTECH. Available at: https://cdn.intechopen.com/pdfs/48191.pdf
- Wikipedia contributors. (2020, April 20). AP site. In Wikipedia, The Free Encyclopedia. Retrieved 18:15, July 23, 2020, from https://en.wikipedia.org/w/index.php?title=AP_site&oldid=952117602
- Wikipedia contributors. (2020, July 4). Benzo(a)pyrene. In Wikipedia, The Free Encyclopedia. Retrieved 06:15, July 24, 2020, from https://en.wikipedia.org/w/index.php?title=Benzo(a)pyrene&oldid=965990545
- Wikipedia contributors. (2020, June 23). Pyrimidine dimer. In Wikipedia, The Free Encyclopedia. Retrieved 06:54, July 24, 2020, from https://en.wikipedia.org/w/index.php?title=Pyrimidine_dimer&oldid=964108515
- Morimoto, S., Tsuda, M., Bunch, H., Sasanuma, H., Ausin, C. and Takeda, S. (2019) Type II DNA Topoisomerases Cause Spontaneous Double-Strand Breaks in Genomic DNA. Genes 10:868. Available at: https://www.researchgate.net/publication/336916880_Type_II_DNA_topoisomerases_cause_spontaneous_double-strand_breaks_in_genomic_DNA/figures?lo=1
- Ding, L., Cao, J., Lin, W., Chen, H., Xiong, X., Ao, H., Yu, M., Lin, J., Cui, Q. (2020) The Roles of the Cyclin-Dependent Kinases in Cell-Cycle Progression and Therapeutic Strategies in Human Breast Cancer. Int. J. Mol Sci 21(6):1960. Available at: https://www.mdpi.com/1422-0067/21/6/1960/htm
- Verma, N., Franchitto, M., Zonfrilli, A., Cialfi, S., Palermo, R., and Talora, C. (2019) DNA Damage Stress: Cui Prodest? Int. J. Mol. Sci. 20(5):1073. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6429504/
- Fukui, K. (2010) DNA Mismatch Repair in Eukaryotes and Bacteria. J. Nuc. Acids 260512. Available at: https://www.hindawi.com/journals/jna/2010/260512/#copyright
- Petruseva, I.O., Evdokimov, A.N., and Lavrik, O.I. (2014) Molecular Mechanism of Global Genome Nucleotide Excision Repair. Acta Naturae 6(1):23-34. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3999463/
- Decottingnies, A. (2013) Alternative end-joining mechanisms: A historical perspective. Frontiers in Genetics 4(48):48. Available at: https://www.researchgate.net/publication/236129718_Alternative_end-joining_mechanisms_A_historical_perspective
- Vitor, A.C., Huertas, P., Legube, G., and de Almeida, S.F. (2020) Studying DNA Double-Strand Break Repair: An Every-Growing Toolbox. Front. Mol Biosci 7:24. Available at: https://www.frontiersin.org/articles/10.3389/fmolb.2020.00024/full
- Krajewska, M., Fehrmann, R.S.N., de Vries, E.G.E., and van Vugt, A.A.T.M. (2015) Regulators of homologous recombination repair as novel targets for cancer treatment. Front. Genet. 6:96. Available at: https://www.frontiersin.org/articles/10.3389/fgene.2015.00096/full