Home » Student Resources » Online Chemistry Textbooks » CH450 and CH451: Biochemistry - Defining Life at the Molecular Level » Chapter 5: Investigating DNA
MenuCH450 and CH451: Biochemistry - Defining Life at the Molecular Level
Chapter 5: Investigating DNA
5.1 DNA Isolation, Sequencing, and Synthesis
5.2 Bioinformatics
5.3 Cloning and Recombinant Expression
5.4 Microarrays
5.5 In Situ Hybridization
5.6 References
5.1 DNA Isolation, Sequencing, and Synthesis
Genomic DNA vs Complimentary DNA
Genomic deoxyribonucleic acid is chromosomal DNA, in contrast to extra-chromosomal DNA such as that found in the mitochondria of mammals or plasmid structures in bacteria (Figure 5.1). Plasmids will be discussed in more detail in section 5.3 during the discussion of gene cloning and expression. It is also then abbreviated as gDNA. Most organisms have the same genomic DNA in every cell; however, only certain genes are active in each cell to allow for cell function and differentiation within the body.
The genome of an organism (encoded by the genomic DNA) is the (biological) information of heredity which is passed from one generation of organism to the next. That genome is transcribed to produce various RNAs, which are necessary for the function of the organism. Precursor mRNA (pre-mRNA) is transcribed by RNA polymerase II in the nucleus. pre-mRNA is then processed by splicing to remove introns, leaving the exons in the mature messenger RNA (mRNA). Additional processing includes the addition of a 5′ cap and a poly(A) tail to the pre-mRNA. The mature mRNA may then be transported to the cytosol and translated by the ribosome into a protein. Other types of RNA include ribosomal RNA (rRNA) and transfer RNA (tRNA). These types are transcribed by RNA polymerase II and RNA polymerase III, respectively, and are essential for protein synthesis. However 5s rRNA is the only rRNA which is transcribed by RNA Polymerase III.
In genetics, complementary DNA (cDNA) is DNA synthesized from a single-stranded RNA (e.g., messenger RNA (mRNA) or microRNA) template in a reaction catalyzed by the enzyme reverse transcriptase (Figure 5.1). Reverse transcriptase is an enzyme found in retroviruses such as HIV that have RNA as their core genetic material. Upon entering the host cell the RNA is reverse transcribed to produce a copy of cDNA that can then integrate into the hosts genomic DNA. In biotechnology, reverse transcriptase is often used to create cDNA from the mRNA expressed in specific cells or tissues. In this way, the eukaryotic genes can be cloned without any introns housed in the structure. This is especially useful if the goal is to express the protein in a prokaryotic (bacterial) host. Recall that bacterial DNA does not house any intron sequences within its chromosomal DNA. Thus, if you are using a prokaryotic system to express eukaryotic proteins, you must use cDNA, as the prokaryotic system will not be able to remove intron sequences following gene transcription.
The term cDNA is also used, typically in a bioinformatics context, to refer to an mRNA transcript’s sequence, expressed as DNA bases (GCAT) rather than RNA bases (GCAU).
- cDNA is derived from mRNA, so it contains only exons but no introns.
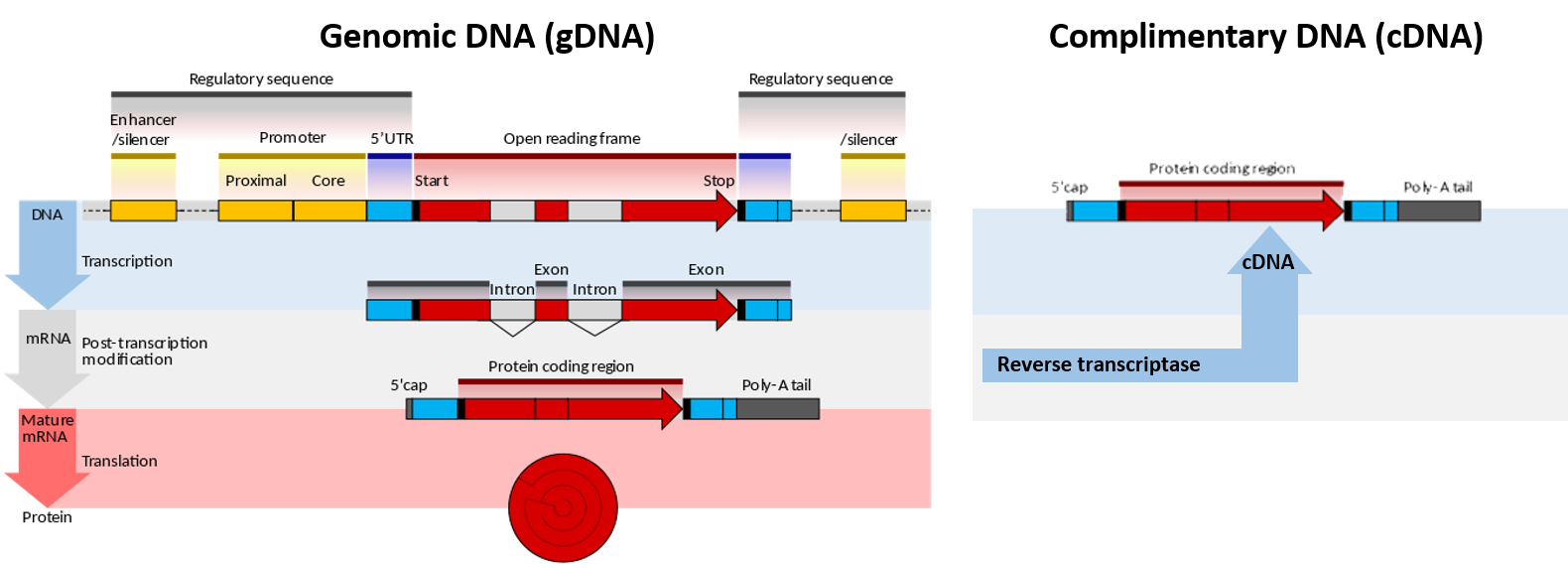
Figure 5.1. Genomic DNA (gDNA) vs Complimentary DNA (cDNA). Lefthand Diagram shows the processing of genomic DNA within a cell to produce a protein (Upper blue panel shows the structural elements common to eukaryotic genes. The process of gene transcription produces a messenger RNA (mRNA) molecule that must be modified post-translationally, gray panel, to remove the non-coding intron sequences and add the 5′-CAP and Poly-A-Tail sections. The mature mRNA is transported from the nucleus to the cytoplasm where it is translated by the ribosome into the protein sequence, red panel.) Righthand Diagram shows that the isolation of mRNA from a cell can be used to synthesize cDNA using the enzyme reverse transcriptase. The resulting cDNA only contains elements from the mature mRNA, including the exons and poly-A tail.
Image modified from Wikipedia
DNA Isolation Techniques
DNA isolation is a process of purification of DNA from sample using a combination of physical and chemical methods. The first isolation of DNA was done in 1869 by Friedrich Miescher. Currently it is a routine procedure in molecular biology or forensic analyses. For the chemical method, there are many different kits used for extraction, and selecting the correct one will save time on kit optimization and extraction procedures. PCR sensitivity detection is considered to show the variation between the commercial kits.
There are three standard DNA purification techniques described below:
- Cells which are to be studied need to be collected.
- Breaking the cell membranes open to expose the DNA along with the cytoplasm within (cell lysis).
- Lipids from the cell membrane and the nucleus are broken down with detergents and surfactants.
- Breaking proteins by adding a protease (optional).
- Breaking RNA by adding an RNase (optional).
- The solution is treated with concentrated salt solution (saline) to make debris such as broken proteins, lipids and RNA to clump together.
- Centrifugation of the solution, which separates the clumped cellular debris from the DNA.
- DNA purification from detergents, proteins, salts and reagents used during cell lysis step. The most commonly used procedures are:
- Ethanol precipitation usually by ice-cold ethanol or isopropanol. Since DNA is insoluble in these alcohols, it will aggregate together, giving a pellet upon centrifugation. Precipitation of DNA is improved by increasing of ionic strength, usually by adding sodium acetate.
- Phenol–chloroform extraction in which phenol denatures proteins in the sample. After centrifugation of the sample, denaturated proteins stay in the organic phase while aqueous phase containing nucleic acid is mixed with the chloroform that removes phenol residues from solution.
- Minicolumn purification that relies on the fact that the nucleic acids may bind (adsorption) to the solid phase (silica or other) depending on the pH and the salt concentration of the buffer (Figure 5.2).
Cellular and histone proteins bound to the DNA can be removed either by adding a protease or by having precipitated the proteins with sodium or ammonium acetate, or extracted them with a phenol-chloroform mixture prior to the DNA-precipitation.
After isolation, the DNA is dissolved in slightly alkaline buffer, usually in a Tris-EDTA buffer, or in ultra-pure water. Modifications made to these standard techniques are often done if the tissue being used is difficult to breakdown, if contaminants persist in the lysis solution that inhibit further reactions, or if the sample is extremely minimal, as is often the case in forensic investigations. In addition, different commercial kits will be tailored for the isolation of larger genomic DNA or smaller plasmid DNA.

Figure 5.2 Silica Spin Column Used for DNA Purification. Spin column-based nucleic acid purification is a solid phase extraction method to quickly purify nucleic acids. This method relies on the fact that nucleic acid will bind to the solid phase of silica under certain conditions and then released when those conditions are altered. For binding, a buffer solution is added to the DNA lysate along with ethanol or isopropanol. This forms the binding solution. The binding solution is transferred to a spin column and the column is put in a centrifuge. The centrifuge forces the binding solution through a silica gel membrane that is inside the spin column. If the pH and salt concentration of the binding solution are optimal, the nucleic acid will bind to the silica gel membrane as the solution passes through. To wash non-specific cellular components from the column, the flow-through is removed and a wash buffer is added to the column. The column is put in a centrifuge again, forcing the wash buffer through the membrane. This removes any remaining impurities from the membrane, leaving only the nucleic acid bound to the silica gel. To elute, the wash buffer is removed and a low salt elution buffer (or simply water) is added to the column. The column is put in a centrifuge again, forcing the elution buffer through the membrane. The elution buffer displaces the nucleic acid from the column allowing it to be collected in the flow through. Unlike RNA which degrades very quickly, DNA is quite stable and can be stored for long periods of time at -20oC.
Image by Squidonius
DNA Sequencing Techniques
DNA sequencing is the process of determining the nucleic acid sequence – the order of nucleotides in DNA. It includes any method or technology that is used to determine the order of the four bases: adenine, guanine, cytosine, and thymine. The advent of rapid DNA sequencing methods has greatly accelerated biological and medical research and discovery.
Knowledge of DNA sequences has become indispensable for basic biological research, and in numerous applied fields such as medical diagnosis, biotechnology, forensic biology, virology and biological systematics. Comparing healthy and mutated DNA sequences can diagnose different diseases including various cancers, characterize antibody repertoire, and can be used to guide patient treatment. Having a quick way to sequence DNA allows for faster and more individualized medical care to be administered, and for more organisms to be identified and cataloged.
The rapid speed of sequencing attained with modern DNA sequencing technology has been instrumental in the sequencing of complete DNA sequences, or genomes, of numerous types and species of life, including the human genome and other complete DNA sequences of many animal, plant, and microbial species.
The first DNA sequences were obtained in the early 1970s by academic researchers using laborious methods based on two-dimensional chromatography. Following the development of fluorescence-based sequencing methods with a DNA sequencer, DNA sequencing has become easier and orders of magnitude faster.
The canonical structure of DNA has four bases: thymine (T), adenine (A), cytosine (C), and guanine (G). DNA sequencing is the determination of the physical order of these bases in a molecule of DNA. However, DNA bases are often modified by epigenetic processes to control gene expression. Thus, many other modified bases that may be present in a DNA molecule than the standard four bases. For example, in some viruses (specifically, bacteriophage), cytosine may be replaced by hydroxymethyl- or hydroxymethylglucose cytosine. In eukaryotic DNA, variant bases with methyl groups or phosphosulfate may be found (Figure 5.3). Depending on the sequencing technique, a particular modification, e.g., the 5mC (5 -methylcytosine) common in humans, may or may not be detected.
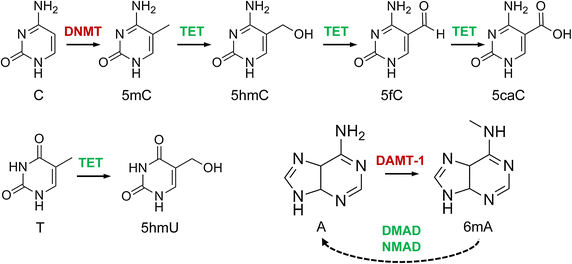
Figure 5.3 DNA modifications with epigenetic regulatory functions and their interdependencies. Cytosine (C) is methylated to 5-methylcytosine (5mC) by DNA methyltransferases (DNMT) and then further oxidized to 5hmC, 5fC and 5caC by Tet dioxygenases. 5-Hydroxyuracil (5hmU) is produced by Tet-catalysed oxidation of thymine (T). N6-methyladenine (6mA) is likely catalysed by DNA N6 adenine methyltransferases (DAMT-1 in C. elegans), even though the biochemical activity of these enzymes remains to be characterized. The Tet-like ALKB enzymes NMAD (N6-methyl adenine demethylase 1) and DMAD (DNA 6mA demethylase) have been shown to be involved in 6mA demethylation in C. elegans and in Drosophila, respectively, possibly by using a conserved dioxygenase mechanism.
Image by Breiling and Lyko (2015) Epigenetics and Chromatin 8:24
-
Early DNA sequencing methods
The first method for determining DNA sequences involved a location-specific primer extension strategy established by Ray Wu at Cornell University in 1970. DNA polymerase catalysis and specific nucleotide labeling, both of which figure prominently in current sequencing schemes, were used to sequence the cohesive ends of lambda phage DNA. Between 1970 and 1973, Wu, R Padmanabhan and colleagues demonstrated that this method can be employed to determine any DNA sequence using synthetic location-specific primers. Frederick Sanger then adopted this primer-extension strategy to develop more rapid DNA sequencing methods at the MRC Centre, Cambridge, UK and published a method for “DNA sequencing with chain-terminating inhibitors” in 1977. Walter Gilbert and Allan Maxam at Harvard also developed sequencing methods, including one for “DNA sequencing by chemical degradation”. In 1973, Gilbert and Maxam reported the sequence of 24 basepairs using a method known as wandering-spot analysis. Advancements in sequencing were aided by the concurrent development of recombinant DNA technology, allowing DNA samples to be isolated from sources other than viruses.
Maxam-Gilbert sequencing requires radioactive labeling at one 5′ end of the DNA and purification of the DNA fragment to be sequenced. Chemical treatment then generates breaks at a small proportion of one or two of the four nucleotide bases in each of four reactions (G, A+G, C, C+T). The concentration of the modifying chemicals is controlled to introduce on average one modification per DNA molecule. Thus a series of labeled fragments is generated, from the radiolabeled end to the first “cut” site in each molecule. The fragments in the four reactions are electrophoresed side by side in denaturing acrylamide gels for size separation. To visualize the fragments, the gel is exposed to X-ray film for autoradiography, yielding a series of dark bands each corresponding to a radiolabeled DNA fragment, from which the sequence may be inferred.
The technical aspects of Maxam-Gilbert sequencing caused it to go out of favor once the Sanger sequencing method had been well established, as described below.
-
Sanger Sequencing Method
The chain-termination method developed by Frederick Sanger and coworkers in 1977 soon became the method of choice, owing to its relative ease and reliability. When invented, the chain-terminator method used fewer toxic chemicals and lower amounts of radioactivity than the Maxam-Gilbert method. Because of its comparative ease, the Sanger method was soon automated and was the method used in the first generation of DNA sequencers.
The classical chain-termination method requires a single-stranded DNA template, a DNA primer, a DNA polymerase, normal deoxynucleotidetriphosphates (dNTPs), and modified di-deoxynucleotidetriphosphates (ddNTPs), the latter of which terminate DNA strand elongation. These chain-terminating nucleotides lack a 3′-OH group required for the formation of a phosphodiester bond between two nucleotides, causing DNA polymerase to cease extension of DNA when a modified ddNTP is incorporated. The ddNTPs may be radioactively or fluorescently labelled for detection in automated sequencing machines.
The DNA sample is divided into four separate sequencing reactions, containing all four of the standard deoxynucleotides (dATP, dGTP, dCTP and dTTP) and the DNA polymerase. To each reaction is added only one of the four dideoxynucleotides (ddATP, ddGTP, ddCTP, or ddTTP), while the other added nucleotides are ordinary ones (Figure 5.4).
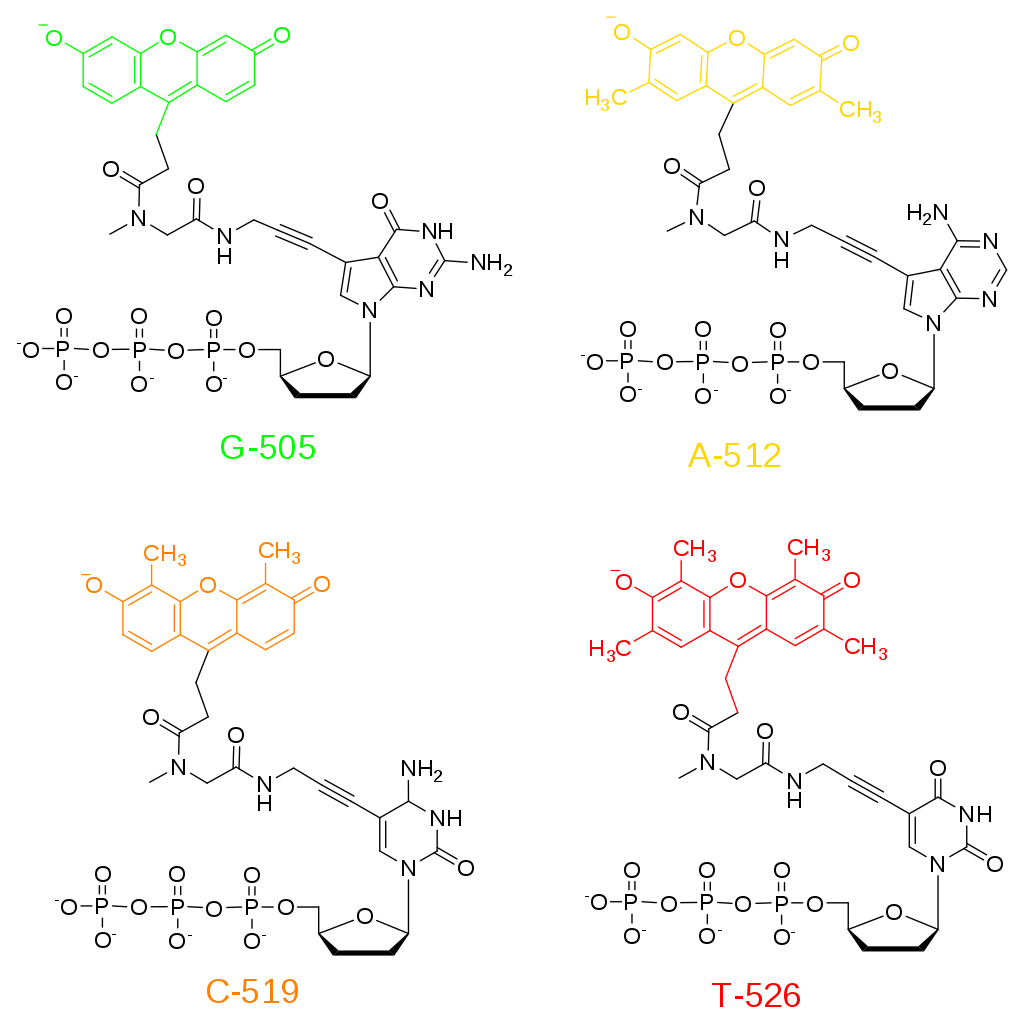
Figure 5.4. Fluorescent ddNTPs for Sanger Sequencing. Dideoxynucleotides are utilized for sequencing as they cannot be extended further once they are incorporated into the nacent DNA.
Image by Fibonachi
The dideoxynucleotide concentration should be approximately 100-fold lower than that of the corresponding deoxynucleotide (e.g. 0.005mM ddTTP : 0.5mM dTTP) to allow enough fragments to be produced while still transcribing the complete sequence. In total, four separate reactions are needed in this process to test all four ddNTPs (Figure 5.5).
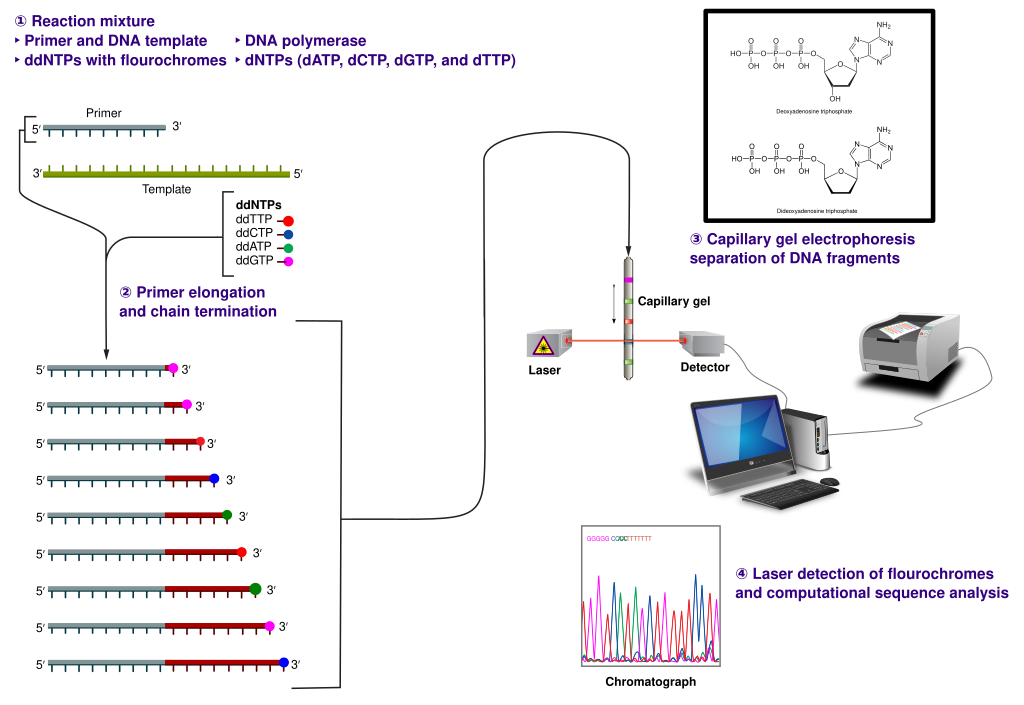
Figure 5.5. The Sanger (chain-termination) method for DNA sequencing. (1) A primer is annealed to a sequence, (2) Reagents are added to the primer and template, including: DNA polymerase, dNTPs, and a small amount of all four dideoxynucleotides (ddNTPs) labeled with fluorophores. During primer elongation, the random insertion of a ddNTP instead of a dNTP terminates synthesis of the chain because DNA polymerase cannot react with the missing hydroxyl. This produces all possible lengths of chains. (3) The products are separated on a single lane capillary gel, where the resulting bands are read by a imaging system. (4) This produces several hundred thousand nucleotides a day, data which require storage and subsequent computational analysis.
Image by Estevezj
Following rounds of template DNA extension from the bound primer, the resulting DNA fragments are heat denatured and separated by size using gel electrophoresis. This technique was frequently performed using a denaturing polyacrylamide-urea gel with each of the four reactions run in one of four individual lanes (lanes A, T, G, C). The DNA bands may then be visualized by autoradiography or UV light and the DNA sequence can be directly read off the X-ray film or gel image (Figure 5.6).

Figure 5.6. Traditional Sanger Sequencing Gel. Sequence visualized by autoradiography. Each lane contains a single reactions that has all four regular nucleotides and a small amount of one of the dideoxynucleotides (ddNTPs). Overtime, the ddNTPs will be incorporated at each position containing that specific nucleotide. The gel can then be read from the bottom to the top, as the smallest fragments (those fragments terminated the closest to the primer at the 5′-end) will run the farthest distance in the gel. The sequence of this fragment is:
5′-TACGAGATATATGGCGTTAATACGATATATTGGAACTTCTATTGC-3′
Image by John Schmidt
Automation of the Sanger sequencing method was made possible when the shift from radioactively tagged nucleotides to fluorescently tagged nucleotides was made. Within the automated sequencers, capillary gel electrophoresis is performed rather than separating the samples using gel electrophoresis. The output from capillary electrophoresis are fluorescent peak trace chromatograms (Figure 5.7). Automated DNA-sequencing instruments (DNA sequencers) can sequence up to 384 DNA samples in a single batch. Batch runs may occur up to 24 times a day greatly enhancing the speed with which samples may be sequenced and analyzed. Common challenges of DNA sequencing with the Sanger method include poor quality in the first 15-40 bases of the sequence due to primer binding and deteriorating quality of sequencing traces after 400-500 bases.

Figure 5.7 Side by Side Comparison of Gel Electrophoresis and Capillary Electrophoresis. Lefthand Diagram shows the traditional autoradiogram of Sanger sequencing samples. The Righthand Diagram shows the same reactions using fluorescently tagged ddNTPs separated by capillary electrophoresis. The chromatogram output is shown on the far right.
Image by Abizar
Sanger sequencing is the method which prevailed from the 1980s until ~2005. Over that period, great advances were made in the technique, such as fluorescent labeling, capillary electrophoresis, and general automation. These developments allowed much more efficient sequencing, leading to lower costs. The Sanger method, in mass production form, is the technology which produced the first human genome in 2001, ushering in the age of genomics.
-
Microfluidic Sanger Sequencing
Microfluidic Sanger sequencing is a lab-on-a-chip application for DNA sequencing, in which the Sanger sequencing steps (thermal cycling, sample purification, and capillary electrophoresis) are integrated on a wafer-scale chip using nanoliter-scale sample volumes (Figure 5.8). This technology generates long and accurate sequence reads, while obviating many of the significant shortcomings of the conventional Sanger method (e.g. high consumption of expensive reagents, reliance on expensive equipment, personnel-intensive manipulations, etc.) by integrating and automating the Sanger sequencing steps.

Figure 5.8 Lab-On-A-Chip Technologies. Example of a microfluidic lab-on-a chip-device sitting on a polystyrene dish. Stainless steel needles inserted into the device serve as access points for fluids into small channels within the device, which are about the size of a human hair.
Image by National Institute of Standards and Technology (NIST)
-
Next Generation Sequencing
Next-generation sequencing (NGS), also known as high-throughput sequencing, is the catch-all term used to describe a number of different modern sequencing technologies. These technologies allow for sequencing of DNA and RNA much more quickly and cheaply than the previously used Sanger sequencing, and as such revolutionized the study of genomics and molecular biology. Such technologies include:
Illumina Sequencing – In NGS, vast numbers of short reads are sequenced in a single stroke using the lab-on-a-chip technology described above. To do this, the input sample must be cleaved into short sections. In Illumina sequencing, 100-150bp reads are used. Somewhat longer fragments are ligated to generic adaptors and annealed to a slide using the adaptors. PCR is carried out to amplify each read, creating a spot with many copies of the same read. They are then separated into single stranded DNA to be sequenced (Figure 5.9).
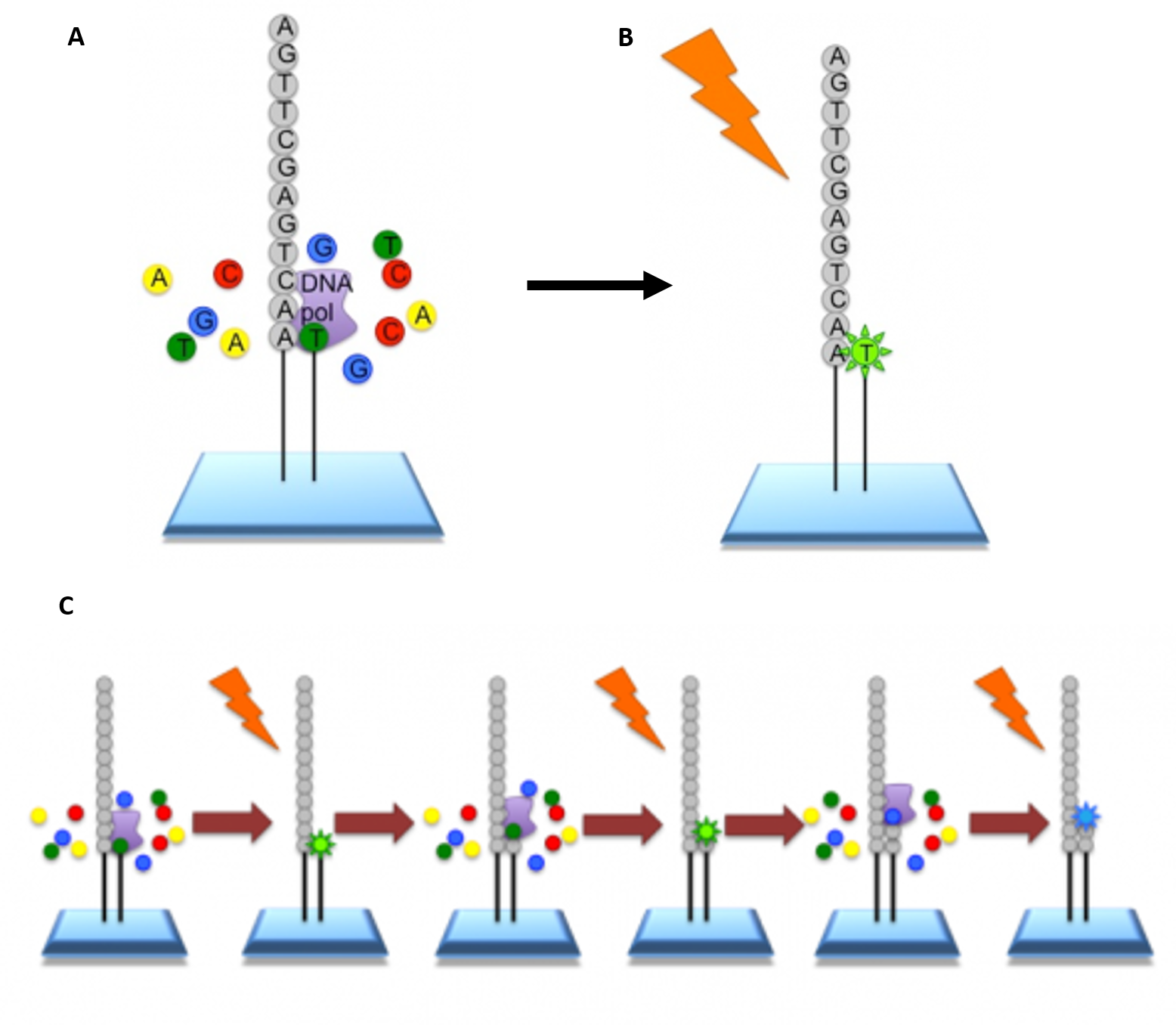
Figure 5.9 Procedure for Illumina Sequencing. (A) The slide with PCR amplified fragments of DNA is flooded with nucleotides and DNA polymerase. These nucleotides are fluorescently labelled with each color corresponding to a specific base. The reactions also have a terminator present, so that only one base is added at a time. (B) An image is taken of the slide. In each reaction location, there will be a fluorescent signal indicating that a specific base that has been added. (C) The data is recorded and the slide is then prepared for the next cycle. In preparation, the terminators are removed, which will allow the next base to be added, and the fluorescent signal is cleaved, preventing the fluorescent signal from contaminating the next image. The process is repeated, adding one nucleotide at a time (G, A, T, or C) and imaging in between. All of the sequence reads will be the same length as single bases are added at each cycle.
Image modified from EMBL-EBI
Roche 454-Sequencing is similar to the Illumina process but can sequence much longer reads. Like Illumina, it does this by sequencing multiple reads at once by reading optical signals as bases are added.
As in Illumina, the DNA or RNA is fragmented into shorter reads, in this case up to 1kb (1,000bp). Generic adaptors are added to the ends and these are annealed to beads, one DNA fragment per bead. The fragments are then amplified by PCR using adaptor-specifc primers. Each bead is then placed in a single well of a slide. So each well will contain a single bead, covered in many PCR copies of a single sequence. The wells also contain DNA polymerase and sequencing buffers (Figure 5.10).
5.10 Procedure for Roche 454 Sequencing. (A) Once the PCR product is attached to the bead, the slide is flooded with one of the four NTP species. Where this nucleotide is next in the sequence, it is added to the sequence read. If that single base repeats, then more will be added. So if we flood with Guanine bases, and the next in the sequence is G, one G will be added, however if the next part of the sequence is GGGG, then four Gs will be added. (B) The addition of each nucleotide releases a light signal. These locations of signals are detected and used to determine which beads the nucleotides are added to. (C) The NTP mix is washed away. The next NTP mix is now added and the process repeated, cycling through the four NTPs. All of the sequence reads from 454 sequencing will be different lengths, because different numbers of bases will be added with each cycle.
Image modified by EMBL-EBI
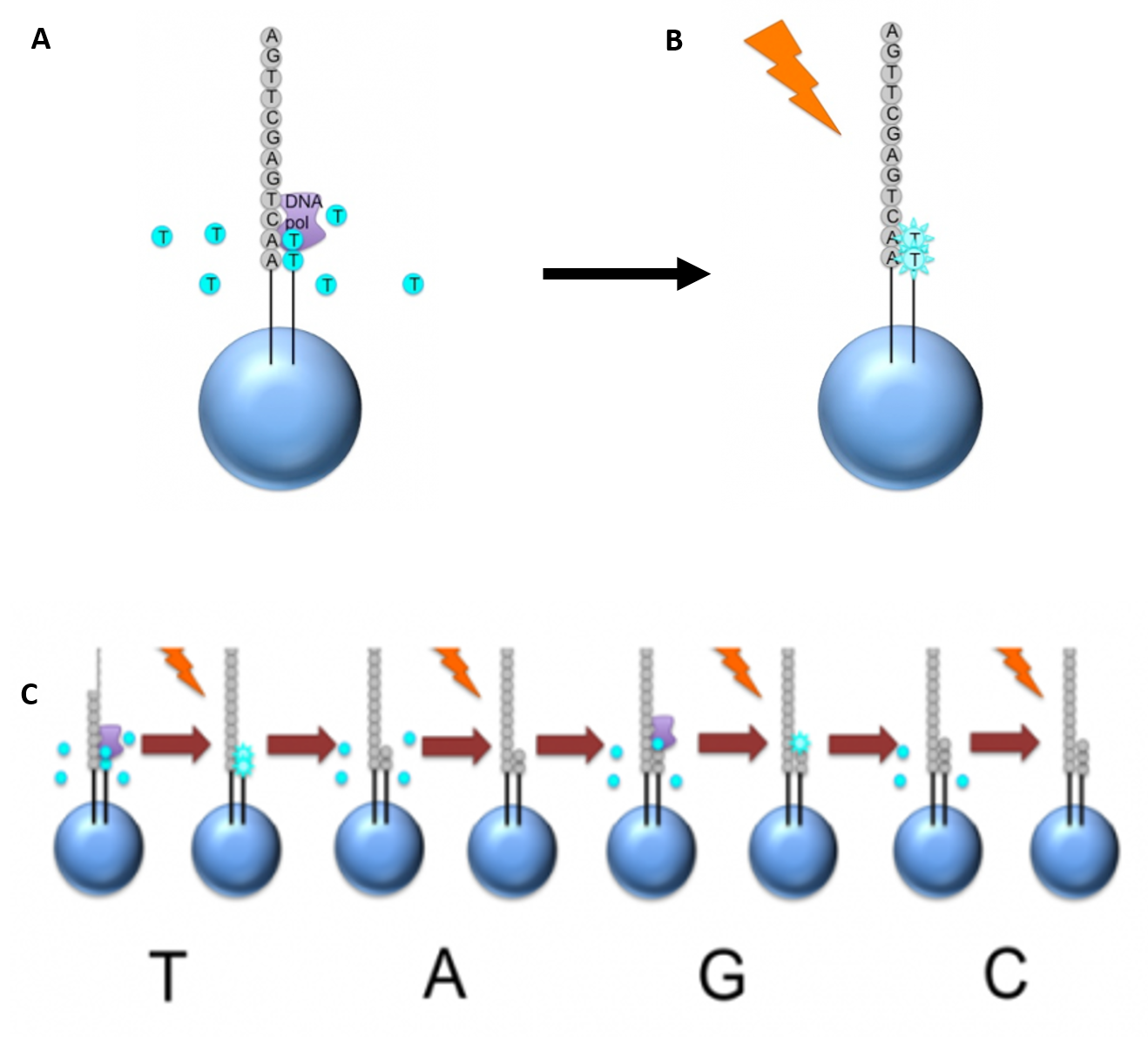
Newer technologies such as the Ion Torrent Technology and the MinION System detect sequence data using electrical signals on a semiconductor chip, rather than optically reading dye-labeled nucleotides. This is possible as the addition of a dNTP to the DNA polymer causes the release of an H+ ion (Figure 5.11). As in other kinds of NGS, the input DNA or RNA is fragmented, this time ~200bp. Adaptors are added and one molecule is placed onto a bead. The molecules are amplified on the bead by emulsion PCR. Each bead is placed into a single well of a slide.
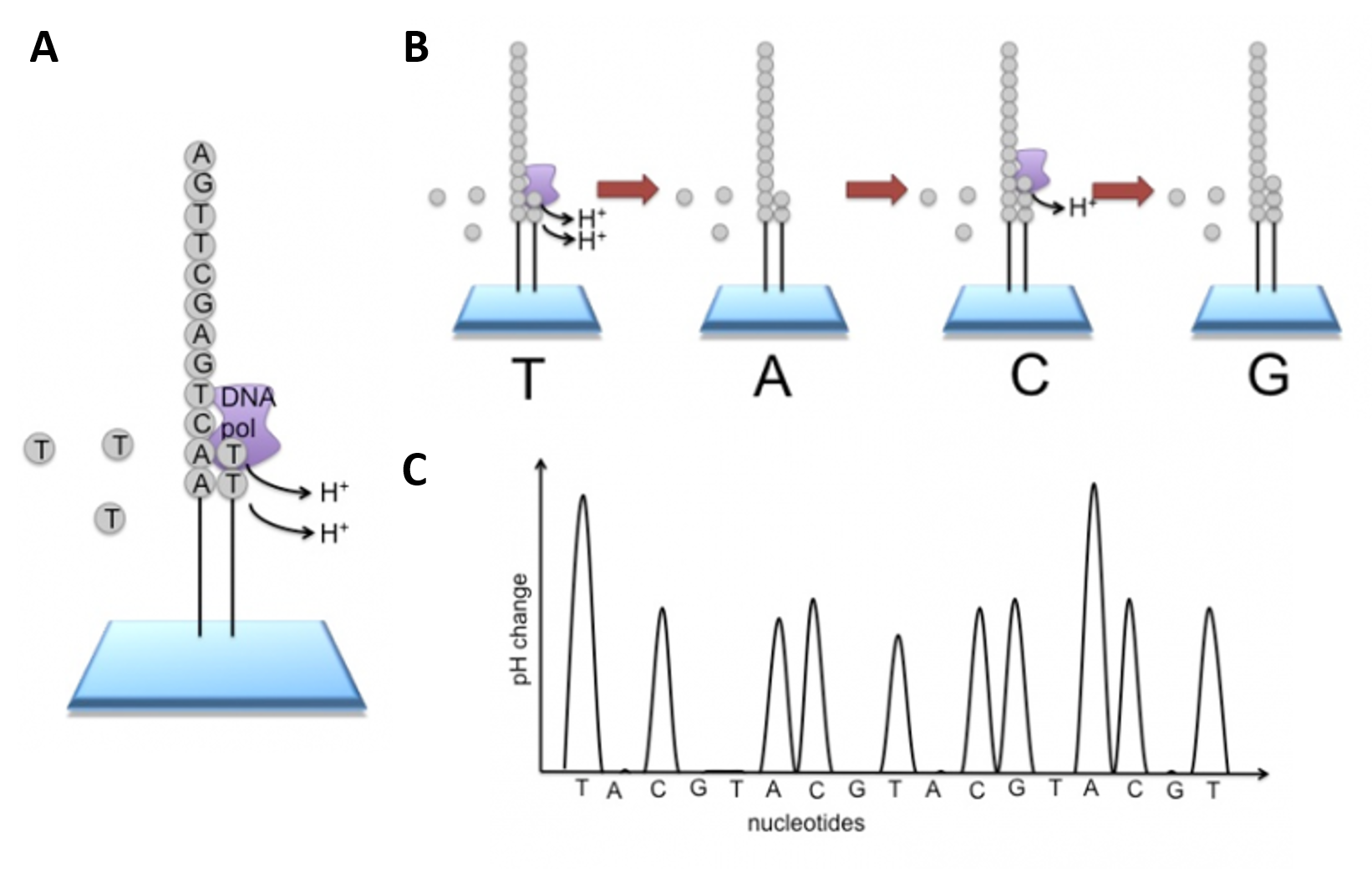
Figure 5.11 Ion Torrent Sequencing Technology. (A) Similar to 454 sequencing, the slide is flooded with a single species of dNTP, along with buffers and polymerase. The pH is monitored in each of the wells after the addition of the specific dNTP. The pH will decrease when the dNTP is incorporated into the polymer causing the release of a proton (H+). The changes in pH allow us to determine if that base and how many thereof, were added to the sequence read. (B) The dNTPs are washed away and the process is repeated cycling through the different dNTP species. (C) The pH change, if any, is used to determine how many bases (if any) were added with each cycle.
Image modified from EMBL-EBI
Such ion technologies that do not require optical detection, have enabled the production of small hand-held DNA sequencing devices that can be plugged into the USB drive on a laptop computer and utilized in the field under real time collection conditions (Figure 5.12).


Figure 5.12 The MinION Portable Real-Time Sequencing Device. The MinION can produce up to 30 Gb of DNA sequence data per sample
Image by Oxford Nanopore Technologies
The four main advantages of NGS over classical Sanger sequencing are:
Sample size
NGS is significantly cheaper, quicker, needs significantly less DNA and is more accurate and reliable than Sanger sequencing. Let us look at this more closely. For Sanger sequencing, a large amount of template DNA is needed for each read. Several strands of template DNA are needed for each base being sequenced (i.e. for a 100bp sequence you’d need many hundreds of copies, for a 1000bp sequence you’d need many thousands of copies), as a strand that terminates on each base is needed to construct a full sequence. In NGS, a sequence can be obtained from a single strand. In both kinds of sequencing multiple staggered copies are taken for contig construction and sequence validation.
Speed
NGS is quicker than Sanger sequencing in two ways. Firstly, the chemical reaction may be combined with the signal detection in some versions of NGS, whereas in Sanger sequencing these are two separate processes. Secondly and more significantly, only one read (maximum ~1kb) can be taken at a time in Sanger sequencing, whereas NGS is massively parallel, allowing 300Gb of DNA to be read on a single run on a single chip.
Cost
The reduced time, manpower and reagents in NGS mean that the costs are much lower. The first human genome sequence cost in the region of $2.7 billion in 2003. Using modern Sanger sequencing methods, aided by data from the known sequence, a full human genome still cost $300,000 in 2006. Sequencing a human genome with NGS today costs roughly $1,000.
Accuracy
Repeats are intrinsic to NGS, as each read is amplified before sequencing, and because it relies on many short overlapping reads, so each section of DNA or RNA is sequenced multiple times. Also, because it is so much quicker and cheaper, it is possible to do more repeats than with Sanger sequencing. More repeats means greater coverage, which leads to a more accurate and reliable sequence, even if individual reads are less accurate for NGS.
Sanger sequencing can be used to give much longer sequence reads. However, the parallel nature of NGS means that longer reads can be constructed from many contiguous short reads.
DNA Synthesis Techniques
DNA synthesis is the natural or artificial creation of deoxyribonucleic acid (DNA) molecules. The term DNA synthesis can refer to DNA replication (which will be covered in more detail in Chapter XX), polymerase chain reaction (PCR) or gene synthesis (physically creating artificial gene sequences).
-
Polymerase Chain Reaction (PCR)
Polymerase chain reaction (PCR) refers to a technique employed widely in the basic and biomedical sciences. PCR is a laboratory technique utilized to amplify specific segments of DNA for a wide range of laboratory and/or clinical applications. Building on the work of Panet and Khorana’s successful amplification of DNA in-vitro, Kary Mullis and coworkers developed PCR in the early 1980s, having been met with a Nobel prize only a decade later. Allowing for more than the billion-fold amplification of specific target regions, it has become instrumental in many applications including the cloning of genes, the diagnosis of infectious diseases, and the screening of prenatal infants for deleterious genetic abnormalities.
Fundamentals
The main components of PCR are a template, primers, free nucleotide bases, and the DNA polymerase enzyme. The DNA template contains the specific region that you wish to amplify, such as the DNA extracted from a piece of hair for example. Primers, or oligonucleotides, are short strands of single-stranded DNA complementary to the 3′ end of each target region. Both a forward and a reverse primer are required, one for each complementary strand of DNA. DNA polymerase is the enzyme that carries out DNA replication. Thermostable analogues of DNA polymerase I, such as Taq polymerase, which was originally found in a bacterium that grows in hot springs, is a common choice due to its resistance to the heating and cooling cycles necessary for PCR.
PCR takes advantage of the complementary base pairing, double-stranded nature, and melting temperature of DNA molecules. This process involves cycling through 3 sequential rounds of temperature dependent reactions: DNA melting (denaturation), annealing and enzyme-driven DNA replication (elongation). Denaturation begins by heating the reaction to about 95oC, disrupting the hydrogen bonds that hold the two strands of template DNA together. Next, the reaction is reduced to around 50 to 65oC, depending on the physicochemical variables of the primers, enabling annealing of complementary base pairs. The primers, which are added to the solution in excess, bind to the beginning of the 3′ end of each template strand and prevent re-hybridization of the template strand with itself. Lastly, enzyme-driven DNA replication, or elongation, begins by setting the reaction temperature to the amount which optimizes the activity of DNA polymerase, which is around 75 to 80oC. At this point, DNA polymerase, which needs double-stranded DNA to begin replication, synthesizes a new DNA strand by assembling free-nucleotides in solution in the 3′ to 5′ direction to produce 2 full sets of complementary strands. The newly synthesized DNA is now identical to the template strand and will be used as such in the progressive PCR cycles (Video 5.1).
Video 5.1: Polymerase Chain Reaction (PCR). (1) Steps of Traditional PCR Process. (2) Non-specific fluorophore detection in qPCR, and (3) Specific hybridization probe detection in qPCR.
Video by Mousa Ghannam
Given that previously synthesized DNA strands serve as templates, the amplification of DNA using PCR increases at an exponential rate, where the copies of DNA double at the end of each replication step. The exponential replication of the target DNA eventually plateaus around 30 to 40 cycles mainly due to reagent limitation, but can also be due to inhibitors of the polymerase reaction found in the sample, self-annealing of the accumulating product, and accumulation of pyrophosphate molecules.
Real-Time PCR
At its advent, PCR technology was limited to qualitative and or semi-quantitative analysis due to limitations on the ability to quantitate nucleic acids. At that time, to verify if the target gene was amplified successfully, the DNA product was separated by size via agarose gel electrophoresis. Ethidium bromide, a molecule that fluoresces when bound to dsDNA, could give a rough estimate of DNA amount by roughly comparing the brightness of separated bands, but was not sensitive enough for rigorous quantitative analysis.
Improvements in fluorophore development and instrumentation led to thermocyclers that no longer required measurement of only end-product DNA. This process, known as real-time PCR, or quantitative PCR (qPCR), has allowed for the detection of dsDNA during amplification. qPCR thermocyclers are equipped with the ability to excite fluorophores at specific wavelengths, detect their emission with a photodetector, and record the values. The sensitive collection of numerical values during amplification has strongly enhanced quantitative analytical power.
There are two main types of fluorophores used in qPCR: those that bind specifically to a given target sequence and those that do not. The sensitivity of fluorophores has been an important aspect of qPCR development. One of the most effective and widely used non-specific markers, SYBR Green, after binding to the minor groove of dsDNA, exhibits a 1000-fold increase in fluorescence compared to being free in solution (Video 5.1). However, if even more specificity is desired, a sequence-specific oligonucleotide, or hybridization probe, can be added, which binds to the target gene at some point in front of the primer (after the 3′ end). These hybridization probes contain a reporter molecule at the 5′ end and a quencher molecule at the 3′ end. The quencher molecule effectively inhibits the reporter from fluorescing while the probe is intact. However, upon contact with DNA polymerase I, the hybridization probe is cleaved, allowing for the fluorescence of the dye (Video 5.1).
Reverse-Transcription PCR
Since its advent, PCR technology has been creatively expanded upon, and reverse-transcription PCR (RT-PCR) is one of the most important advances. Real-time PCR is frequently confused with reverse-transcription PCR, but they are separate techniques. In RT-PCR, the DNA amplified is derived from mRNA by using reverse-transcriptase enzymes, to produce a cDNA copy of the gene. Using primers sequences for genes of interest, traditional PCR methods can be used with the cDNA to study the expression of genes qualitatively. Currently, reverse-transcription PCR is commonly used with real-time PCR, which allows one to quantitatively measure the relative change in gene expression across different samples.
Issues of Concern
One disadvantage of PCR technology is that it is extremely sensitive. Trace amounts of RNA or DNA contamination in the sample can produce extremely misleading results. Another disadvantage is that the primers designed for PCR requires sequence data, and therefore can only be used to identify the presence or absence of a known pathogen or gene. Another limitation is that sometimes the primers used for PCR can anneal non-specifically to sequences that are similar, but not identical, to the target gene.
Another potential issue of using PCR is the possibility of primer dimer (PD) formation. PD is a potential by-product and consists of primer molecules that have hybridized to each other due to the strings of complementary bases in the primers. The DNA polymerase amplifies the PD, leading to competition for PCR reagents that could be used to amplify the target sequences.
Clinical Significance
PCR amplification is an indispensable tool with various applications within medicine. Often, it is used to test for the presence of specific alleles, such as in the case of prospective parents screening for genetic carriers, but it can also be used to diagnose the presence of disease directly and for mutations in the developing embryo. For example, the first time PCR was used in this way was for the diagnosis of sickle cell anemia through the detection of a single gene mutation.
Additionally, PCR has greatly revolutionized the diagnostic potential for infectious diseases, as it can be used to rapidly determine the identity of microbes that were traditionally unable to be cultured, or that required weeks for growth. Pathogens routinely detected using PCR include Mycobacterium tuberculosis, human immunodeficiency virus, herpes simplex virus, syphilis, and countless other pathogens. Moreover, qPCR is not only used for testing the qualitative presence of microbes but also to quantify the bacterial, fungal, and viral loads.
The sensitivity of diagnostic tools for mutations to oncogenes and tumor suppression genes has been improved at least 10,000 fold due to PCR, allowing for earlier diagnosis of cancers like leukemia. PCR has also enabled more nuanced and individualized therapies for cancer patients. Additionally, PCR can be used for the tissue typing done that is vital to organ implantation and has even been proposed as a replacement for antibody-based tests for blood type. PCR also has clinical applications in the field of prenatal testing for various genetic diseases and/or clinical pathologies. Samples are obtained either via amniocentesis or chorionic villus sampling.
In forensic medicine, short pieces of repeating, highly polymorphic DNA, coined short tandem repeats (STRs), are amplified and used to compare specific variation within genes to differentiate individuals.[9] Primers specific for the loci of these STRs are used and amplified using PCR. Various loci contain STRs in the human genome, and the statistical power of this technique is enhanced by checking multiple sites.
-
Gene Synthesis
Artificial gene synthesis, sometimes known as DNA printing is a method in synthetic biology that is used to create artificial genes in the laboratory. Based on solid-phase DNA synthesis, it differs from molecular cloning and polymerase chain reaction (PCR) in that it does not have to begin with preexisting DNA sequences. Therefore, it is possible to make a completely synthetic double-stranded DNA molecule with no apparent limits on either nucleotide sequence or size.
The method has been used to generate functional bacterial or yeast chromosomes containing approximately one million base pairs. Creating novel nucleobase pairs in addition to the two base pairs in nature could greatly expand the genetic code.
Synthesis of the first complete gene, a yeast tRNA, was demonstrated by Har Gobind Khorana and coworkers in 1972. Synthesis of the first peptide- and protein-coding genes was performed in the laboratories of Herbert Boyer and Alexander Markham, respectively.
Commercial gene synthesis services are now available. Approaches are most often based on a combination of organic chemistry and molecular biology techniques and entire genes may be synthesized “de novo”, without the need for template DNA. Gene synthesis is an important tool in many fields of recombinant DNA technology including heterologous gene expression, vaccine development, gene therapy and molecular engineering. The synthesis of nucleic acid sequences can be more economical than classical cloning and mutagenesis procedures. It is also a powerful and flexible engineering tool for creating and designing new DNA sequences and protein functions.
Gene optimization
While the ability to make increasingly long stretches of DNA efficiently and at lower prices is a technological driver of this field, increasingly attention is being focused on improving the design of genes for specific purposes. Early in the genome sequencing era, gene synthesis was used as an (expensive) source of cDNAs that were predicted by genomic or partial cDNA information but were difficult to clone. As higher quality sources of sequence verified cloned cDNA have become available, this practice has become less urgent.
Producing large amounts of protein from gene sequences (or at least the protein coding regions of genes, the open reading frame) found in nature can sometimes prove difficult and is a problem of sufficient impact that scientific conferences have been devoted to the topic. Many of the most interesting proteins sought by molecular biologists are normally regulated to be expressed in very low amounts in wild type cells. Redesigning these genes offers a means to improve gene expression in many cases. Rewriting the open reading frame is possible because of the degeneracy of the genetic code. Thus it is possible to change up to about a third of the nucleotides in an open reading frame and still produce the same protein. The available number of alternate designs possible for a given protein is astronomical. For a typical protein sequence of 300 amino acids there are over 10150 codon combinations that will encode an identical protein. Codon optimization, or replacing rarely used codons with more common codons sometimes have dramatic effects. Further optimizations such as removing RNA secondary structures can also be included. At least in the case of E. coli, protein expression is maximized by predominantly using codons corresponding to tRNA that retain amino acid charging during starvation. Computer programs written to perform these, and other simultaneous optimizations are used to handle the enormous complexity of the task. A well optimized gene can improve protein expression 2 to 10 fold, and in some cases more than 100 fold improvements have been reported. Because of the large numbers of nucleotide changes made to the original DNA sequence, the only practical way to create the newly designed genes is to use gene synthesis.
Oligonucleotides are chemically synthesized using building blocks called nucleoside phosphoramidites. These can be normal or modified nucleosides which have protecting groups to prevent their amines, hydroxyl groups and phosphate groups from interacting incorrectly. One phosphoramidite is added at a time, the 5′ hydroxyl group is deprotected and a new base is added and so on. The chain grows in the 3′ to 5′ direction, which is backwards relative to DNA biosynthesis in vivo. At the end, all the protecting groups are removed.

Figure 5.13 Four-step phosphoramidite oligodeoxynucleotide synthesis cycle. The phosphoramidite method, pioneered by Marvin Caruthers in the early 1980s, and enhanced by the application of solid-phase technology and automation, is now firmly established as the method of choice. Phosphoramidite oligonucleotide synthesis proceeds in the 3 to 5 direction (opposite to the 5 to 3 direction of DNA biosynthesis in DNA replication). One nucleotide is added per synthesis cycle. The phosphoramidite DNA synthesis cycle consists of a series of steps outlined in the figure
Image by Ni, S., et al (2017) International Journal of Molecular Sciences 18(8):1683
Nevertheless, being a chemical process, several incorrect interactions occur leading to some defective products. The longer the oligonucleotide sequence that is being synthesized, the more defects there are, thus this process is only practical for producing short sequences of nucleotides. The current practical limit is about 200 bp (base pairs) for an oligonucleotide with sufficient quality to be used directly for a biological application. HPLC can be used to isolate products with the proper sequence. Meanwhile, a large number of oligos can be synthesized in parallel on gene chips. For optimal performance in subsequent gene synthesis procedures they should be prepared individually and in larger scales.
-
DNA synthesis and synthetic biology
The significant drop in cost of gene synthesis in recent years due to increasing competition of companies providing this service has led to the ability to produce entire bacterial plasmids that have never existed in nature. The field of synthetic biology utilizes the technology to produce synthetic biological circuits, which are stretches of DNA manipulated to change gene expression within cells and cause the cell to produce a desired product.
The ability to synthetically produce DNA will enable the development of environmental, medical, and commercially relevant products. For example, in 2015, Novartis, in collaboration with the Synthetic Genomics Vaccines inc. and the US Biomedical Advanced Research and Development Authority, announced that they had effectively created a synthetic DNA influenza vaccine. New synthetic DNA vaccines hold promise to provide an alternative to current egg-produced conventional vaccines that can be plagued by low efficacy.
DNA vaccines are able to avoid many issues associated with egg-based vaccine production by generating viral proteins within host cells. To create a DNA vaccine, an antigen-encoding gene is cloned into a non-replicative expression plasmid, which is delivered to the host by traditional vaccination routes. Host cells which take up the plasmid express the vaccine antigen which can be presented to immune cells via the major histocompatibility complex (MHC) pathways. CD4+ T helper cell activation following MHC class II presentation of secreted DNA vaccine protein is critical for the production of antigen-specific antibodies (Figure 5.14).

Figure 5.14 Creation of a DNA Vaccine. An antigenic gene is synthesized and cloned into a plasmid vector. (Steps regarding the cloning process are described in more detail in section 5.3). The DNA vaccine is delivered to the host, where it will be expressed to produce and present antigen to the host immune system.
After two decades of research, DNA vaccine technology is gaining maturity—several veterinary DNA vaccines are currently licensed for West Nile virus and melanoma, and significantly, the first commercial DNA vaccine against H5N1 in chickens has recently been conditionally approved by the USDA. In addition, ongoing large animal trials of DNA vaccines against other diseases such as against HIV, hepatitis, and Zika virus offer valuable insights that can be applied to influenza DNA vaccine design. Promising approaches have arisen from the numerous studies evaluating different DNA vaccine formulations and delivery systems, but a strategy that consistently elicits protection against influenza in large animal models has not yet emerged. Successful plasmid delivery and the use of appropriate adjuvants remain key challenges that need to be addressed before influenza DNA vaccines become effective for human use.
5.2 Bioinformatics
An unprecedented revolution has been observed in science with recent technological advances, which have provided a large amount of “omic” data. The crescent generation and availability of this information available in public databases were, and still are, a challenge for professionals from different areas. However, what is the challenge? In biology, the main challenge is to make sense of the enormous amount of structural data and sequences that have been generated at multiple levels of biological systems. Still, in bioinformatics, development of tools is necessary (statistical and computational) capable of assisting in understanding the mechanisms underlying biological questions in the study. Besides, if we consider the complexity of science, this is a highly reductionist view. The era of a “new biology” emerges accompanied by the birth/development of other sciences, such as bioinformatics and computational biology, which have an integrated interface of molecular biology. Although considered recently, bioinformatics and genomics have evolved interdependently and promoted a historical impact on the available knowledge.
Bioinformatics, a hybrid science that links biological data with techniques for information storage, distribution and analysis to support multiple areas of scientific research including biomedicine. It mainly involving molecular biology and genetics, computer science, mathematics and statistics. Data intensive, large-scale biological problems are addressed from a computational point of view. Bioinformatics is fed by high-throughput data-generating experiments, including genomic sequence determinations and measurements of gene expression patterns. Database projects curate and annotate the data and then distribute it via the World Wide Web. Mining these data leads to scientific discoveries and to the identification of new clinical applications.
A bioinformatics solution usually involves the following steps:
- Collect statistics from biological data
- Build a computational model
- Solve a computational modeling problem
- Test and evaluate a computational algorithm
It also addresses the following aspects:
- Types of biological information and databases
- Sequence analysis and molecular modeling
- Genomic analysis
- Systems biology
In the field of medicine in particular, a number of important applications for bioinformatics have been discovered. For example, it is used to identify correlations between gene sequences and diseases, to predict protein structures from amino acid sequences, to aid in the design of novel drugs, and to tailor treatments to individual patients based on their DNA sequences (pharmacogenomics). In bioinformatics, we can now conduct global analyses of all the available data with the aim of uncovering common principles that apply across many systems and highlight novel features.
Some applications of bioinformatics in biotechnology are given below:
-
Genomics
To manage an escalating amount of genomic information, bioinformatic tools are required to maintain and analyze the DNA sequences from different organism. Determination of sequence homology, gene finding, coding region identification, structural and functional analyses of genomic sequences etc, all this is possible by the use of different bioinformatics tools and software packages.
Given below is a list of few bioinformatics tools used in genomics (Table 5.1).
Table 5.1 Bioinformatics Tools/Databases used in Genomics
| Bioinformatics tools | Purpose |
|---|---|
| Carrie | Transcriptional regulatory networks database |
| CisML | Motif detection tool |
| ICSF | Identification of conserved structural features in TF binding sites |
| oPossum | Tool for motif searching |
| Promoser | Promoter extraction tool from eukaryotic organisms |
| REPFIND | Determine clustered repeats in DNA fragment |
| Cluster‐Buster | Tool for predicting motifs cluster in DNA sequences |
| Cister | Finds regulatory regions in DNA fragments |
| Clover | Find overrepresented motifs in DNA sequences |
| GLAM | Tool for predicting functional motifs |
| MotifViz | Identification of overrepresented motifs |
| NECorr | Tool for analysing gene expression data |
| ROVER | Predicts overrepresented motifs in DNA fragments |
| SeqVISTA | Sequences viewer tool |
| DNADynamo | Tool to find transcription factors with over‐represented binding sites in the upstream regions of co‐expressed human genes |
Table from Kahn, N.T. (2018)
Bioinformatics plays an important role in comparative genomics by determing the genomic structural and functional relationship between different biological species.
Given below is a list of few bioinformatics tools used in comparative genomics (Table 5.2).
Table 5.2 Bioinformatics Tools/Databases used in Comparative Genomics
| Bioinformatics tools | Purpose |
|---|---|
| BLAST | DNA or protein sequence alignment tool |
| HMMER | Homologous protein sequences searching tool |
| Clustal Omega | Multiple sequence alignments tool |
| Sequerome | Sequence profiling tool |
| ProtParam | Predicts the physico-chemical properties of proteins |
| novoSNP | Predicts single point mutation in DNA sequences |
| ORF Finder | Find open reading frame in putative genes |
| Virtual Foorprint | Analysis of whole prokaryotic genome |
| WebGeSTer | Predicts gene termination sites during transcription |
| Genscan | Find exon-intron sites in DNA sequences |
| Softberry Tools | Genomes annotation tool along with the structure and function prediction of biological molecules |
| MEGA | Study evolutionary relationship |
| MOLPHY | Maximum likelihood based phylogenetic analysis tool |
| PHYLIP | Tool for phylogenetic studies |
| JStree | Tool for viewing and editing phylogenetic trees |
| Jalview | It is an alignment editing tool |
| The DNA Data Bank of Japan | Resources for nucleotide sequences |
| Rfam | Database contains collection of RNA families |
| Uniprot | Protein sequence database |
| Protein Data Bank | Database provide data on structures of nucleic acids, proteins etc |
| SWISS PROT | Database containing the manually annotated protein sequences |
| InterPro | Provide information on protein families, its conserved domains and actives sites |
| Proteomics Identifications Database | Contains data on functional characterization and post-translation modification of proteins and peptides |
| Ensembl | Database containing annotated genomes of eukaryotes including human, mouse and other vertebrates |
| Medherb | Database for medicinally herbs |
Table from Kahn, N.T. (2018)
-
Proteomics:
Advanced molecular based techniques led to the accumulation of huge proteomic data of protein activity patterns, interactions, profiling, composition, structural information, image analysis, peptide mass fingerprinting, peptide fragmentation fingerprinting etc. This enormous data could be managed by using different tools of bioinformatics.
Given below is a list of few bioinformatics tools used in proteomics (Table 5.3).
Table 5.3 Bioinformatics Tools/Databases used in Proteomics.
| Bioinformatics tools | Purpose |
|---|---|
| K2 / FAST | Protein structure alignment tool |
| SMM | Tool for determining peptides binding to major histocompatibility complex |
| ZDOCK | Protein‐protein docking tool |
| Docking Benchmark | Tool to evaluate docking algorithms performance |
| ZDOCK Server | An automated server for running ZDOCK |
| MELANIE | Proteomic analysis for analysing 2D-Gel images |
Table from Kahn, N.T. (2018)
Clinical bioinformatics is an emerging new field of bioinformatics that employs various bioinformatics tool such as computer aided drug designing to design novel drugs, vaccines, DNA drug modelling, and in silico drug testing to produce new and effective drugs in a shorter time frame with lower risks.
Bioinformatic tools such as NCI, NCIP (part of NCI) and CBIIT have played an important role in genomics, proteomics, imaging, and metabolomics to increase our knowledge of the molecular basis of cancer.
Using numerous bioinformatics tools, phylogenetic analysis of the molecular data can easily be achieved in a short period of time by constructing phylogenetic trees to study its evolutionary relationship based on sequence alignment.
A number of databases consists of DNA profiles of known delinquents. Advancement in microarray technology, bayesian networks, and programming algorithms provides an effective method of evidence organization and interpretation.
Though bioinformatics has limited impact on forensic since there is a need for more advanced algorithms and computational applications so that the established databases may exhibit interoperability with each other.
Progressions in structural /functional genomics and molecular technologies such as genome sequencing and DNA microarrays generates valuable knowledge which explains nutrition in relation of an individual’s genetics which directly influences its metabolism. Because of the influx of bioinformatics tools, nutrition-related research is tremendously increased.
Regulation of gene expression is the core of functional genomics allowing researchers to apply genomic data to molecular technologies that can quantify the amount of actively transcribing genes in any cell at any time (e.g. gene expression arrays).
Given below is a list of few bioinformatics tools used in gene expression study Table 5.4.
Table 5.4 Bioinformatics Tools/Databases Used in Gene Expression
| Bioinformatics tools | Purpose |
|---|---|
| GeneChords | Conserved gene retrieval tool |
| Bioconductor | Provides tools for the analysis of high throughput genomic data |
| GXD | Gene expression database for the laboratory mouse |
| Inverted Repeats Finder | Find inverted repeats in genomic DNA |
| BU ORChID | Database stores hydroxyl radical cleavage data of DNA sequences |
| ODB | Predicts functional gene clusters |
| RNA Fold Support | Predicts RNA structure based on mutations in alleles |
| CellNetVis | Visualizing tool for biological complexes and networks |
| Tandem Repeat Finder | Finds tandem repeats in genomic DNA |
| VisANT | Tools for visualizing and analysing many biological interactions |
| PROMO | Identification of transcription factor binding sites |
| ConTra V.3 | Detection of transcription factor binding site |
Table remixed from Kahn, N.T. (2018)
New improvements in computing algorithms and available structural simulation databases of recognized structures has brought molecular modeling into conventional food chemistry. Such simulations will make it possible to improve food quality by developing new food additives by comprehending the basis of taste tenacity, antagonism and complementation.
Protein topology prediction is now so much easy thanks to bioinformatics which helps in the prediction of 3D structure of a protein to gain an insight into its function as well.
Given below is a list of few bioinformatics tools used in protein structure and function prediction Table 5.
Table 5.5 Bioinformatics Tools/Databases Used in Protein Structure and Function Prediction
| Bioinformatics tools | Purpose |
|---|---|
| CATH | Tool for the categorized organization of proteins |
| Phyre2 | Tool for protein structure prediction |
| HMMSTR | For the prediction of sequence-structure correlations in proteins |
| MODELLER | Predicts 3D structure of protein |
| JPRED/APSSP2 | Predicts secondary structures of proteins |
| RaptorX | Predicts protein structure |
| QUARK | Predicts Protein Structure |
Table remixed from Kahn, N.T. (2018)
Doctors will be able to analyze a patient’s genetic profile and prescribe the best available drug therapy and dosage from the beginning by employing bioinformatics tool.
Microbes have been studied at very basic level with the help of bioinformatics tools required to analyze their unique set of genes that enables them to survive under unfavorable conditions.
5.3 Cloning and Recombinant Expression
To accomplish the applications described above, biochemists must be able to extract, manipulate, and analyze nucleic acids. To understand the basic techniques used to work with nucleic acids, remember that nucleic acids are macromolecules made of nucleotides (a sugar, a phosphate, and a nitrogenous base). The phosphate groups on these molecules each have a net negative charge. An entire set of DNA molecules in the nucleus of eukaryotic organisms is called the genome. DNA has two complementary strands linked by hydrogen bonds between the paired bases.
Unlike DNA in eukaryotic cells, RNA molecules leave the nucleus. Messenger RNA (mRNA) is analyzed most frequently because it represents the protein-coding genes that are being expressed in the cell.
DNA isolation techniques have been described in section 5.1 and are the first step used to study or manipulate nucleic acids. RNA can also be extracted and is studied to understand gene expression patterns in cells. RNA is naturally very unstable because enzymes that break down RNA are commonly present in nature. Some are even secreted by our own skin and are very difficult to inactivate. During RNA extraction, RNase inhibitors and the special treatment of glassware are used to reduce the risk of destroying the sample during isolation
-
Gel Electrophoresis
Because nucleic acids are negatively charged ions at neutral or alkaline pH in an aqueous environment, they can be moved by an electric field. Gel electrophoresis is a technique used to separate charged molecules on the basis of size and charge. The nucleic acids can be separated as whole chromosomes or as fragments. The nucleic acids are loaded into a slot at one end of a gel matrix, an electric current is applied, and negatively charged molecules are pulled toward the opposite end of the gel (the end with the positive electrode). Smaller molecules move through the pores in the gel faster than larger molecules; this difference in the rate of migration separates the fragments on the basis of size. The nucleic acids in a gel matrix are invisible until they are stained with a compound that allows them to be seen, such as a dye. Distinct fragments of nucleic acids appear as bands at specific distances from the top of the gel (the negative electrode end) that are based on their size (Figure 5.15). A mixture of many fragments of varying sizes appear as a long smear, whereas uncut genomic DNA is usually too large to run through the gel and forms a single large band at the top of the gel.
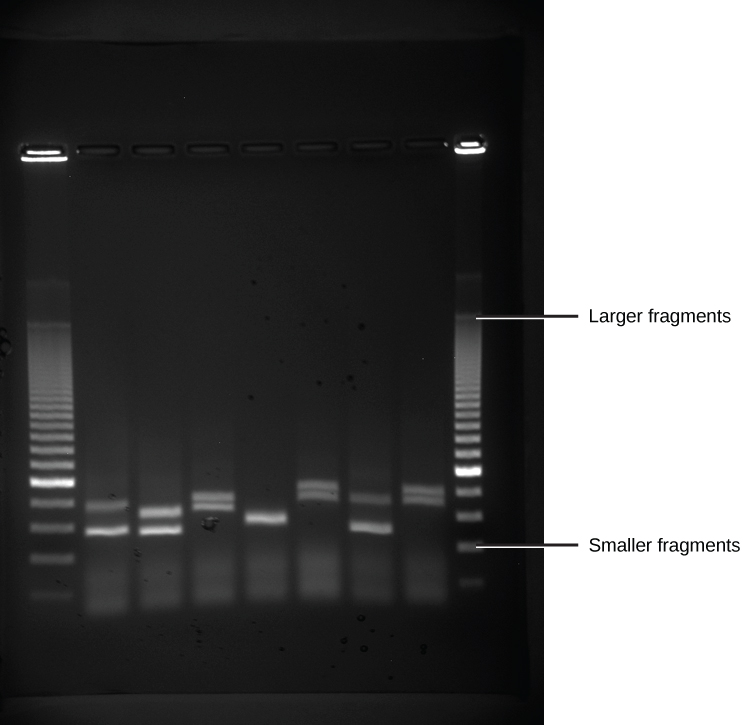
Figure 5.15 DNA Gel Electrophoresis. Shown are DNA fragments from six samples run on a gel, stained with a fluorescent dye and viewed under UV light. (credit: modification of work by James Jacob, Tompkins Cortland Community College)
-
Polymerase Chain Reaction (PCR)
The details of PCR are discussed in section 5.1. This technique is used in DNA cloning to rapidly increase the number of copies of specific regions of DNA.
-
Cloning
In general, cloning means the creation of a perfect replica. Typically, the word is used to describe the creation of a genetically identical copy. In biology, the re-creation of a whole organism is referred to as “reproductive cloning.” Long before attempts were made to clone an entire organism, researchers learned how to copy short stretches of DNA—a process that is referred to as molecular cloning.
Molecular cloning allows for the creation of multiple copies of genes, expression of genes, and study of specific genes. To get the DNA fragment into a bacterial cell in a form that will be copied or expressed, the fragment is first inserted into a cloning vector.
A cloning vector is a small piece of DNA that can be stably maintained in an organism, and into which a foreign DNA fragment can be inserted for cloning purposes. The cloning vector may be DNA taken from a virus, the cell of a higher organism, or it may be the plasmid of a bacterium. The vector therefore contains features that allow for the convenient insertion or removal of a DNA fragment to or from the vector, for example by treating the vector and the foreign DNA with a restriction enzyme that cuts the DNA. DNA fragments thus generated contain either blunt ends or overhangs known as sticky ends, and vector DNA and foreign DNA with compatible ends can then be joined together by molecular ligation. After a DNA fragment has been cloned into a cloning vector, it may be further subcloned into another vector designed for more specific use.
There are many types of cloning vectors, but the most commonly used ones are genetically engineered plasmids. Cloning is generally first performed using Escherichia coli, and cloning vectors in E. coli include plasmids, bacteriophages (such as phage λ), cosmids, and bacterial artificial chromosomes (BACs). Some DNA, however, cannot be stably maintained in E. coli, for example very large DNA fragments. For these studies, other organisms such as yeast may be used. Cloning vectors in yeast include yeast artificial chromosomes (YACs).

Figure 5.16 Example of a Common Cloning Vector.
Image by Ayacop and Yikrazuul
All commonly used cloning vectors in molecular biology have key features necessary for their function, such as a suitable cloning site with restriction enzymes and a selectable marker. Others may have additional features specific to their use. For reasons of ease and convenience, cloning is often performed using E. coli. Thus, the cloning vectors used often have elements necessary for their propagation and maintenance in E. coli, such as a functional origin of replication (ori). The ColE1 origin of replication is found in many plasmids. Some vectors also include elements that allow them to be maintained in another organism in addition to E. coli, and these vectors are called shuttle vectors.
Cloning site
All cloning vectors have features that allow a gene to be conveniently inserted into the vector or removed from it. This may be a multiple cloning site (MCS) or polylinker, which contains many unique restriction sites. The restriction sites in the MCS are first cleaved by restriction enzymes, then a PCR-amplified target gene also digested with the same enzymes is ligated into the vectors using DNA ligase. The target DNA sequence can be inserted into the vector in a specific direction if so desired. The restriction sites may be further used for sub-cloning into another vector if necessary.
Other cloning vectors may use topoisomerase instead of ligase and cloning may be done more rapidly without the need for restriction digest of the vector or insert. In this TOPO cloning method a linearized vector is activated by attaching topoisomerase I to its ends, and this “TOPO-activated” vector may then accept a PCR product by ligating both the 5′ ends of the PCR product, releasing the topoisomerase and forming a circular vector in the process. Another method of cloning without the use of DNA digest and ligase is by DNA recombination, for example as used in the Gateway cloning system. The gene, once cloned into the cloning vector (called entry clone in this method), may be conveniently introduced into a variety of expression vectors by recombination.
Restricition Enzymes
Restriction enzymes (also called restriction endonucleases) recognize specific DNA sequences and cut them in a predictable manner; they are naturally produced by bacteria as a defense mechanism against foreign DNA.
As the name implies, restriction endonucleases (or restriction enzymes) are “restricted” in their ability to cut or digest DNA. The restriction that is useful to biochemists is usually a palindromic DNA sequence. Palindromic sequences are the same sequence forwards and backwards. Some examples of palindromes: RACE CAR, CIVIC, A MAN A PLAN A CANAL PANAMA. With respect to DNA, there are 2 strands that run antiparallelel to each other. Therefore, the reverse complement of one strand is identical to the other.
Like with a word palindrome, this means the DNA palindromic sequence reads the same forward and backward. In most cases, the sequence reads the same forward on one strand and backward on the complementary strand. REs often cut DNA into a staggered pattern. When a staggered cut is made in a sequence, the overhangs are complementary (Figure 5.17).
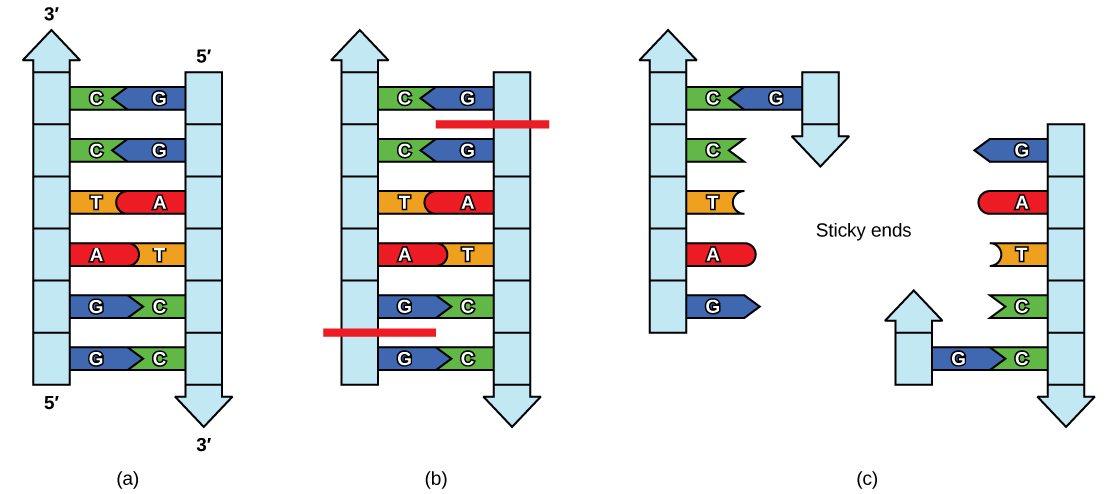
Figure 5.17 Restriction Enzyme Recognition Sequences. In this (a) six-nucleotide restriction enzyme recognition site, notice that the sequence of six nucleotides reads the same in the 5′ to 3′ direction on one strand as it does in the 5′ to 3′ direction on the complementary strand. This is known as a palindrome. (b) The restriction enzyme makes breaks in the DNA strands, and (c) the cut in the DNA results in “sticky ends”. Another piece of DNA cut on either end by the same restriction enzyme could attach to these sticky ends and be inserted into the gap made by this cut.
Molecular biologists also tend to use these special molecular scissors that recognize palindromes of 6 or 8. By using 6-cutters or 8-cutters, the sequences occur throughout large stretches rarely, but often enough to be of utility.



Figure 5.18 Restriction Enzymes. Restriction enzymes recognize palindromic sequences in DNA and hydrolyze covalent phosphodiester bonds of the DNA to leave either “sticky/cohesive” ends or “blunt” ends. This distinction in cutting is important because an EcoRI sticky end can be used to match up a piece of DNA cut with the same enzyme in order to glue or ligate them back together. While endonucleases cut DNA, ligases join them back together. DNA digested with EcoRI can be ligated back together with another piece of DNA digested with EcoRI, but not to a piece digested with SmaI. Another blunt cutter is EcoRV with a recognition sequence of GAT | ATC.
Selectable marker
A selectable marker is carried by the vector to allow the selection of positively transformed cells. Antibiotic resistance is often used as marker, an example being the beta-lactamase gene, which confers resistance to the penicillin group of beta-lactam antibiotics like ampicillin. Some vectors contain two selectable markers, for example the plasmid pACYC177 has both ampicillin and kanamycin resistance gene. Shuttle vectors which are designed to be maintained in two different organisms may also require two selectable markers, although some selectable markers such as resistance to zeocin and hygromycin B are effective in different cell types. Auxotrophic selection markers that allow an auxotrophic organism to grow in minimal growth medium may also be used; examples of these are LEU2 and URA3 which are used with their corresponding auxotrophic strains of yeast.
Another kind of selectable marker allows for the positive selection of plasmid with cloned gene. This may involve the use of a gene lethal to the host cells, such as barnase, Ccda, and the parD/parE toxins. This typically works by disrupting or removing the lethal gene during the cloning process, and unsuccessful clones where the lethal gene still remains intact would kill the host cells, therefore only successful clones are selected.
Reporter genes
Reporter genes are used in some cloning vectors to facilitate the screening of successful clones by using features of these genes that allow successful clone to be easily identified. Such features present in cloning vectors may be the lacZα fragment for α complementation in blue-white selection, and/or marker gene or reporter genes in frame with and flanking the MCS to facilitate the production of fusion proteins. Examples of fusion partners that may be used for screening are the green fluorescent protein (GFP) and luciferase.
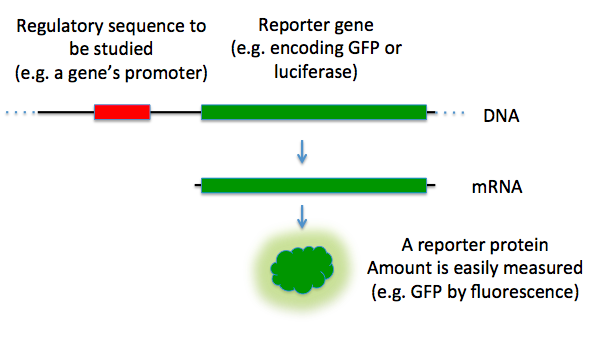
Figure 5.19 Reporter Genes. In this diagram, the green fluorescence protein is used as a reporter gene to study upstream regulatory sequences.
Image by TransControl
If the expression of the targeted gene is desired, then a cloning vector also needs to contain suitable elements for the expression of the cloned target gene, including a promoter and ribosomal binding site (RBS). The target DNA may be inserted into a site that is under the control of a particular promoter necessary for the expression of the target gene in the chosen host. Where the promoter is present, the expression of the gene is preferably tightly controlled and inducible so that proteins are only produced when required. Some commonly used promoters are the T7 and lac promoters. The presence of a promoter is necessary when screening techniques such as blue-white selection are used.
Cloning vectors without promoter and RBS for the cloned DNA sequence are sometimes used, for example when cloning genes whose products are toxic to E. coli cells. Promoter and RBS for the cloned DNA sequence are also unnecessary when first making a genomic or cDNA library of clones since the cloned genes are normally subcloned into a more appropriate expression vector if their expression is required.
-
Types of cloning vectors
A large number of cloning vectors are available, and choosing the right vector may depend a number of factors, such as the size of the insert, copy number and cloning method. Large DNA inserts may not be stably maintained in a general cloning vector, especially for those with a high copy number, therefore cloning large fragments may require more specialized cloning vector.
Plasmids are autonomously replicating circular extra-chromosomal DNA. They are the standard cloning vectors and the ones most commonly used. Most general plasmids may be used to clone DNA insert of up to 15 kb in size. Many plasmids have high copy number, for example pUC19 which has a copy number of 500-700 copies per cell, and high copy number is useful as it produces greater yield of recombinant plasmid for subsequent manipulation. However low-copy-number plasmids may be preferably used in certain circumstances, for example, when the protein from the cloned gene is toxic to the cells.
Bacteriophage
The bacteriophages most commonly used for cloning are the lambda (λ) phage and M13 phage. There is an upper limit on the amount of DNA that can be packed into a phage (a maximum of 53 kb). The average lambda phage genome is roughly 48.5 kb (Figure 5.20). Therefore to allow foreign DNA to be inserted into phage DNA, phage cloning vectors may need to have some of their non-essential genes deleted to make room for the foreign DNA.
There is also a lower size limit for DNA that can be packed into a phage, and vector DNA that is too small cannot be properly packaged into the phage. This property can be used for selection – vector without insert may be too small, therefore only vectors with insert may be selected for propagation.
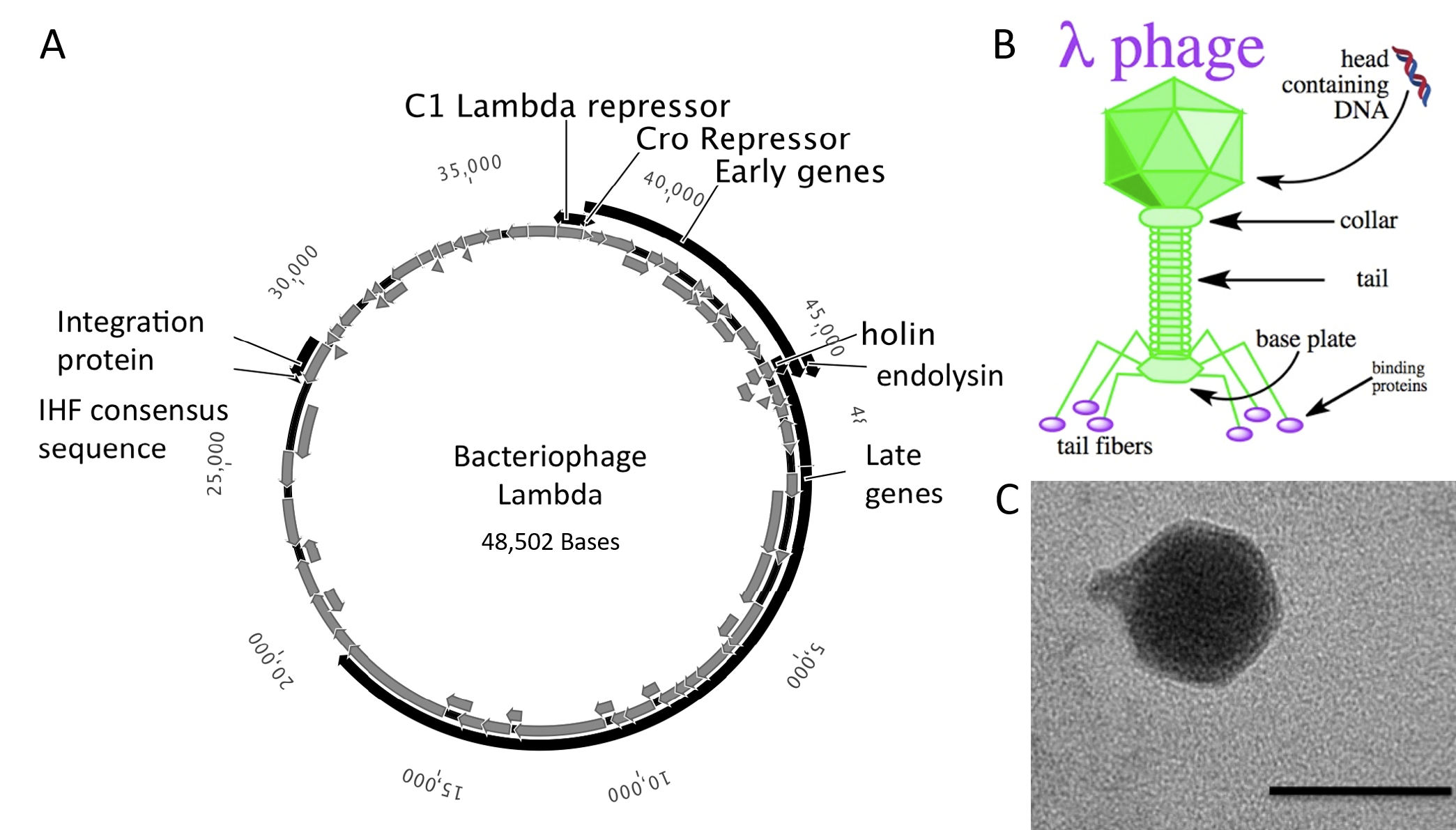
Figure 5.20 Lambda Phage. (A) Schematic representation of the circular genome of the lambda phage (B) Diagram of the Lambda Phage infectious particle and (C) Electron micrograph of the related bacteriophage, vibriophage VvAWI. The bar denotes 50 nm in length.
Images A and C modified from: Nigro, O, Culley, A., and Steward, G.F. (2012) Standards in Genomic Science 6(3):415-26, and image B is from Jack Potte
Cosmid
Cosmids are plasmids that incorporate a segment of bacteriophage λ DNA that has the cohesive end sites (cos) which contains elements required for packaging DNA into λ particles. It is normally used to clone large DNA fragments between 28 and 45 Kb.
Bacterial artificial chromosome
Insert size of up to 350 kb can be cloned in bacterial artificial chromosome (BAC). BACs are maintained in E. coli with a copy number of only 1 per cell. BACs have often been used to sequence the genome of organisms in genome projects, including the Human Genome Project. A short piece of the organism’s DNA is amplified as an insert in BACs, and then sequenced. Finally, the sequenced parts are rearranged in silico, resulting in the genomic sequence of the organism. BACs have largely been replaced in this capacity with faster and less laborious sequencing methods like whole genome shotgun sequencing and now more recently next-gen sequencing.
Yeast artificial chromosome
Yeast artificial chromosome are used as vectors to clone DNA fragments of more than 1 mega base (1Mb = 1000kb = 1,000,000 bases) in size. They are useful in cloning larger DNA fragments as required in mapping genomes such as in human genome project. It contains a telomeric sequence, an autonomously replicating sequence( features required to replicate linear chromosomes in yeast cells). These vectors also contain suitable restriction sites to clone foreign DNA as well as genes to be used as selectable markers.
Human artificial chromosome
Human artificial chromosomes may be potentially useful as a gene transfer vectors for gene delivery into human cells, and a tool for expression studies and determining human chromosome function. It can carry very large DNA fragment (there is no upper limit on size for practical purposes), therefore it does not have the problem of limited cloning capacity of other vectors, and it also avoids possible insertional mutagenesis caused by integration into host chromosomes by viral vector.
Animal and plant viral vectors that infect plant and animal cells have also been manipulated to introduce foreign genes into plant and animal cells. The natural ability of viruses to adsorb to cells , introduce their DNA and replicate have made them ideal vehicles to transfer foreign DNA into eukaryotic cells in culture. A vector based on Simian virus 40 (SV40) was used in first cloning experiment involving mammalian cells. A number of vectors based on other type of viruses like Adenoviruses and Papilloma virus have been used to clone genes in mammals. At present , retroviral vectors are popular for cloning genes in mammalian cells. In case of plant tranformation, viruses including the Cauliflower Mosaic Virus , Tobacco Mosaic Virus and Gemini Viruses have been used with limited success.
-
Summary of DNA Cloning
Figure 5.21 provides a summary of the basic cloning methods most widely used in biochemistry laboratories. Foreign DNA is isolated or amplified using PCR to obtain enough material for the cloning procedure. The DNA is purified and cut with restriction enzymes, and then mixed with a vector that has been cut with the same restriction enzymes. The DNA can then be stitched back together with DNA ligase. The DNA can then be transformed into a host system, often times bacteria, to grow large quantities of the plasmid containing the cloned DNA.
Restriction fragment patterning and DNA sequencing can be used to validate the cloned material.
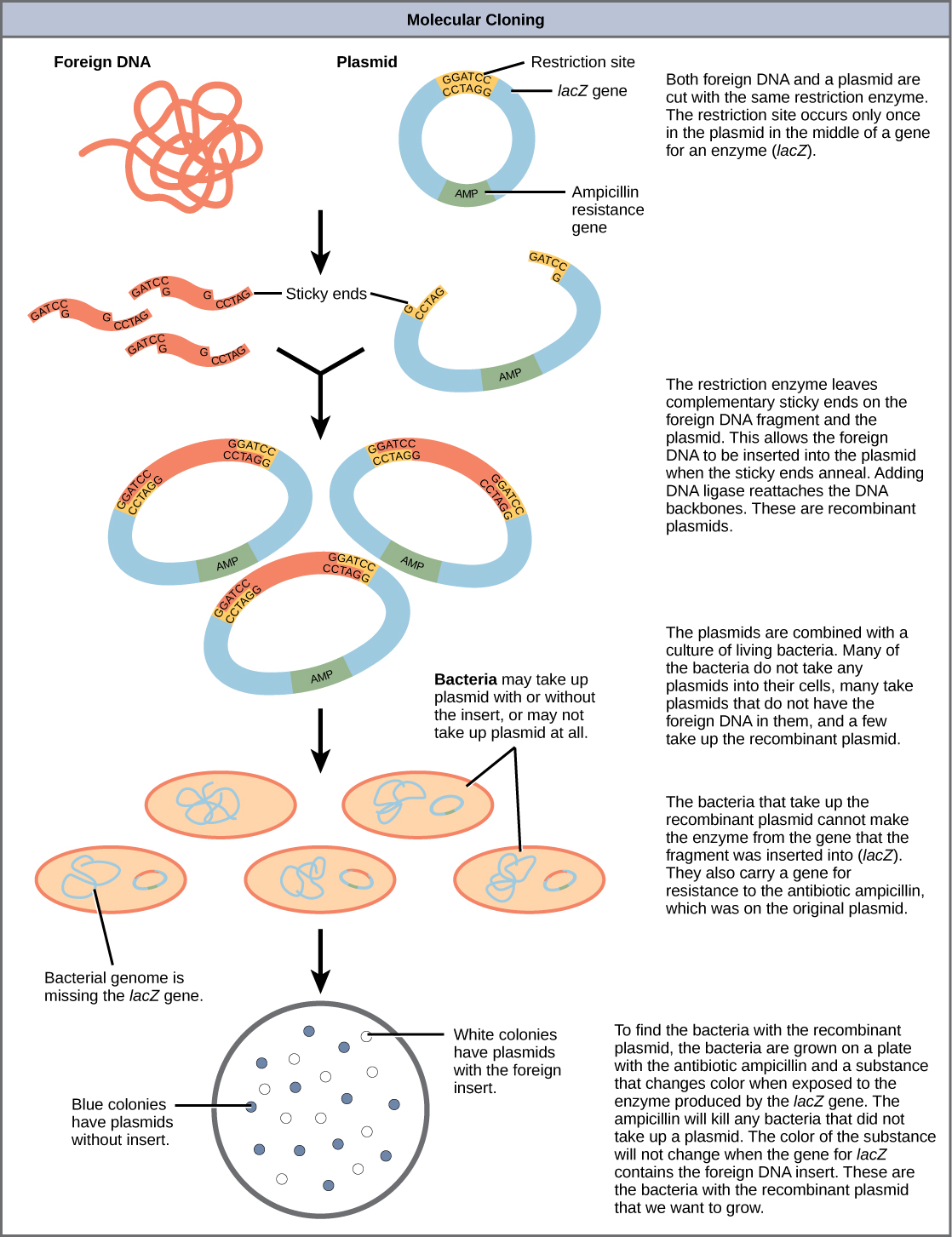
Figure 5.21 Diagram Showing the Major Steps in Cloning.
For a Video Tutorial on DNA Cloning Visit: HHMI – BioInteractive
Plasmids with foreign DNA inserted into them are called recombinant DNA molecules because they contain new combinations of genetic material. Proteins that are produced from recombinant DNA molecules are called recombinant proteins. Not all recombinant plasmids are capable of expressing genes. Plasmids may also be engineered to express proteins only when stimulated by certain environmental factors, so that scientists can control the expression of the recombinant proteins.
-
Reproductive Cloning
Reproductive cloning is a method used to make a clone or an identical copy of an entire multicellular organism. Most multicellular organisms undergo reproduction by sexual means, which involves the contribution of DNA from two individuals (parents), making it impossible to generate an identical copy or a clone of either parent. Recent advances in biotechnology have made it possible to reproductively clone mammals in the laboratory.
Natural sexual reproduction involves the union, during fertilization, of a sperm and an egg. Each of these gametes is haploid, meaning they contain one set of chromosomes in their nuclei. The resulting cell, or zygote, is then diploid and contains two sets of chromosomes. This cell divides mitotically to produce a multicellular organism. However, the union of just any two cells cannot produce a viable zygote; there are components in the cytoplasm of the egg cell that are essential for the early development of the embryo during its first few cell divisions. Without these provisions, there would be no subsequent development. Therefore, to produce a new individual, both a diploid genetic complement and an egg cytoplasm are required. The approach to producing an artificially cloned individual is to take the egg cell of one individual and to remove the haploid nucleus. Then a diploid nucleus from a body cell of a second individual, the donor, is put into the egg cell. The egg is then stimulated to divide so that development proceeds. This sounds simple, but in fact it takes many attempts before each of the steps is completed successfully.
The first cloned agricultural animal was Dolly, a sheep who was born in 1996. The success rate of reproductive cloning at the time was very low. Dolly lived for six years and died of a lung tumor (Figure 5.22). There was speculation that because the cell DNA that gave rise to Dolly came from an older individual, the age of the DNA may have affected her life expectancy. Since Dolly, several species of animals (such as horses, bulls, and goats) have been successfully cloned.
There have been attempts at producing cloned human embryos as sources of embryonic stem cells. In the procedure, the DNA from an adult human is introduced into a human egg cell, which is then stimulated to divide. The technology is similar to the technology that was used to produce Dolly, but the embryo is never implanted into a surrogate mother. The cells produced are called embryonic stem cells because they have the capacity to develop into many different kinds of cells, such as muscle or nerve cells. The stem cells could be used to research and ultimately provide therapeutic applications, such as replacing damaged tissues. The benefit of cloning in this instance is that the cells used to regenerate new tissues would be a perfect match to the donor of the original DNA. For example, a leukemia patient would not require a sibling with a tissue match for a bone-marrow transplant.
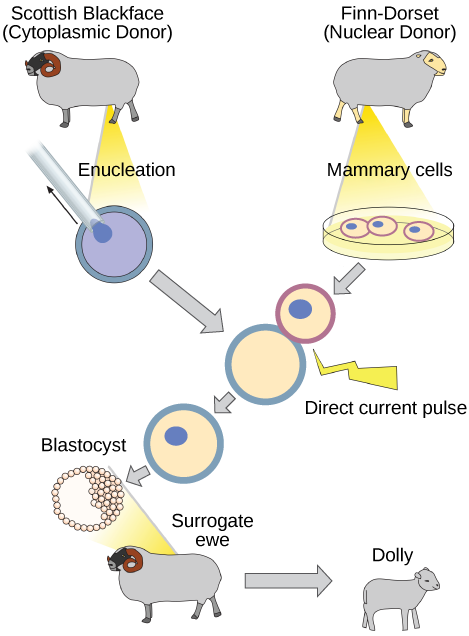
Figure 5.22 Dolly the sheep was the first agricultural animal to be cloned. To create Dolly, the nucleus was removed from a donor egg cell. The enucleated egg was placed next to the other cell, then they were shocked to fuse. They were shocked again to start division. The cells were allowed to divide for several days until an early embryonic stage was reached, before being implanted in a surrogate mother.
Why was Dolly a Finn-Dorset and not a Scottish Blackface sheep?
Because even though the original cell came from a Scottish Blackface sheep and the surrogate mother was a Scottish Blackface, the DNA came from a Finn-Dorset.
-
Genetic Engineering
Using recombinant DNA technology to modify an organism’s DNA to achieve desirable traits is called genetic engineering. Addition of foreign DNA in the form of recombinant DNA vectors that are generated by molecular cloning is the most common method of genetic engineering. An organism that receives the recombinant DNA is called a genetically modified organism (GMO). If the foreign DNA that is introduced comes from a different species, the host organism is called transgenic. Bacteria, plants, and animals have been genetically modified since the early 1970s for academic, medical, agricultural, and industrial purposes.
Watch this short video explaining how scientists create a transgenic animal.
Although the classic methods of studying the function of genes began with a given phenotype and determined the genetic basis of that phenotype, modern techniques allow researchers to start at the DNA sequence level and ask: “What does this gene or DNA element do?” This technique, called reverse genetics, has resulted in reversing the classical genetic methodology. One example of this method is analogous to damaging a body part to determine its function. An insect that loses a wing cannot fly, which means that the wing’s function is flight. The classic genetic method compares insects that cannot fly with insects that can fly, and observes that the non-flying insects have lost wings. Similarly in a reverse genetics approach, mutating or deleting genes provides researchers with clues about gene function. Alternately, reverse genetics can be used to cause a gene to overexpress itself to determine what phenotypic effects may occur.
-
CRISPR Technology
CRISPR stands for clustered regularly interspaced short palindromic repeats and represents a family of DNA sequences found within the genomes of prokaryotic organisms such as bacteria and archaea. These sequences are derived from DNA fragments of bacteriophages that have previously infected the prokaryote and are used to detect and destroy DNA from similar phages during subsequent infections. Hence these sequences play a key role in the antiviral defense system of prokaryotes.

5.23 Crystal structure of a CRISPR RNA-guided surveillance complex, Cascade, bound to a ssDNA target. CRISPR system Cascade protein subunits CasA, CasB, CasC, CasD, and CasE (cyan) bound to CRISPR RNA (green) and viral DNA (red) based on PDB 4QYZ and rendered with PyMOL.
Image from Boghog
Cas9 (or “CRISPR-associated protein 9”) is an enzyme that uses CRISPR sequences as a guide to recognize and cleave specific strands of DNA that are complementary to the CRISPR sequence. Cas9 enzymes together with CRISPR sequences form the basis of a technology known as CRISPR-Cas9 that can be used to edit genes within organisms. This editing process has a wide variety of applications including basic biological research, development of biotechnology products, and treatment of diseases.

The CRISPR-Cas system is a prokaryotic immune system that confers resistance to foreign genetic elements such as those present within plasmids and phages that provides a form of acquired immunity. RNA harboring the spacer sequence helps Cas (CRISPR-associated) proteins recognize and cut foreign pathogenic DNA. Other RNA-guided Cas proteins cut foreign RNA. CRISPR are found in approximately 50% of sequenced bacterial genomes and nearly 90% of sequenced archaea.
5.4 DNA Microarrays
A DNA microarray (also commonly known as DNA chip or biochip) is a collection of microscopic DNA spots attached to a solid surface. Scientists use DNA microarrays to measure the expression levels of large numbers of genes simultaneously or to genotype multiple regions of a genome. Each DNA spot contains picomoles (10−12 moles) of a specific DNA sequence, known as probes (or reporters or oligos). These can be a short section of a gene or other DNA element that are used to hybridize a cDNA or cRNA (also called anti-sense RNA) sample (called target) under high-stringency conditions. Probe-target hybridization is usually detected and quantified by detection of fluorophore-, silver-, or chemiluminescence-labeled targets to determine relative abundance of nucleic acid sequences in the target. The original nucleic acid arrays were macro arrays approximately 9 cm × 12 cm and the first computerized image based analysis was published in 1981. It was invented by Patrick O. Brown.

Figure 5.25 Schematic of DNA Microarrays. Within the organisms, genes are transcribed and spliced to produce mature mRNA transcripts (red). The mRNA is extracted from the organism and reverse transcriptase is used to copy the mRNA into stable ds-cDNA (blue). In microarrays, the ds-cDNA is fragmented and fluorescently labelled (orange). The labelled fragments bind to an ordered array of complementary oligonucleotides, and measurement of fluorescent intensity across the array indicates the abundance of a predetermined set of sequences. These sequences are typically specifically chosen to report on genes of interest within the organism’s genome.
Image by Thomas Shafee
The core principle behind microarrays is hybridization between two DNA strands, the property of complementary nucleic acid sequences to specifically pair with each other by forming hydrogen bonds between complementary nucleotide base pairs. A high number of complementary base pairs in a nucleotide sequence means tighter non-covalent bonding between the two strands. After washing off non-specific bonding sequences, only strongly paired strands will remain hybridized. Fluorescently labeled target sequences that bind to a probe sequence generate a signal that depends on the hybridization conditions (such as temperature), and washing after hybridization. Total strength of the signal, from a spot (feature), depends upon the amount of target sample binding to the probes present on that spot. Microarrays use relative quantitation in which the intensity of a feature is compared to the intensity of the same feature under a different condition, and the identity of the feature is known by its position.
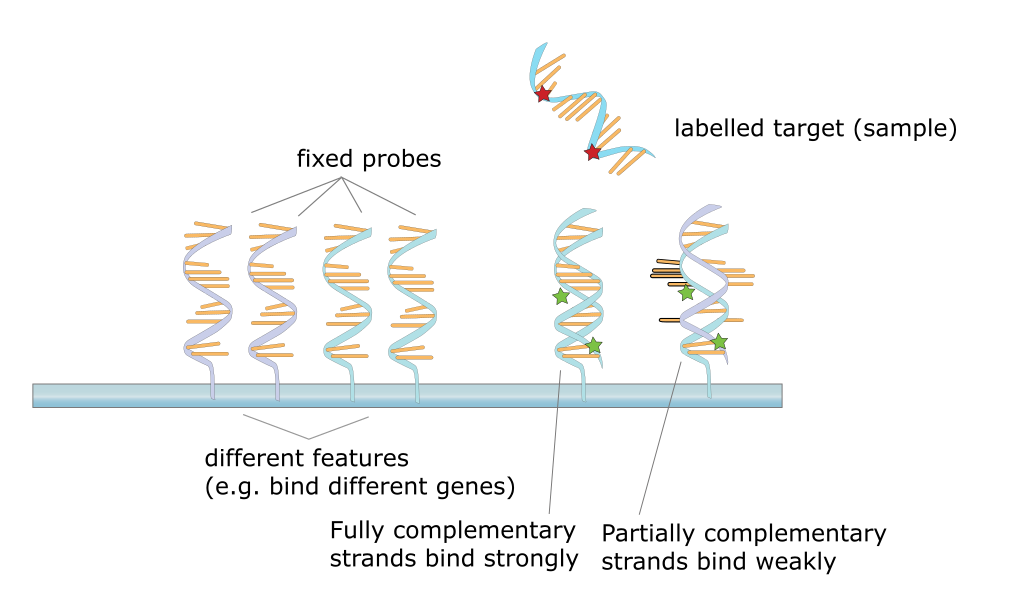
Figure 5.26 Hybridization of Target DNA with the Probe DNA During Microarray Analysis
Image by Squidonius
Many types of arrays exist and the broadest distinction is whether they are spatially arranged on a surface or on coded beads:
- The traditional solid-phase array is a collection of orderly microscopic “spots”, called features, each with thousands of identical and specific probes attached to a solid surface, such as glass, plastic or silicon biochip (commonly known as a genome chip, DNA chip or gene array). Thousands of these features can be placed in known locations on a single DNA microarray.
- The alternative bead array is a collection of microscopic polystyrene beads, each with a specific probe and a ratio of two or more dyes, which do not interfere with the fluorescent dyes used on the target sequence.
DNA microarrays can be used to detect DNA (as in comparative genomic hybridization), or detect RNA (most commonly as cDNA after reverse transcription) that may or may not be translated into proteins. The process of measuring gene expression via cDNA is called expression analysis or expression profiling.
-
Fabrication
Microarrays can be manufactured in different ways, depending on the number of probes under examination, costs, customization requirements, and the type of scientific question being asked. Arrays from commercial vendors may have as few as 10 probes or as many as 5 million or more micrometre-scale probes.
-
Spotted vs. in situ synthesised arrays
Microarrays can be fabricated using a variety of technologies, including printing with fine-pointed pins onto glass slides, photolithography using pre-made masks, photolithography using dynamic micromirror devices, ink-jet printing, or electrochemistry on microelectrode arrays.
In spotted microarrays, the probes are oligonucleotides, cDNA or small fragments of PCR products that correspond to mRNAs. The probes are synthesized prior to deposition on the array surface and are then “spotted” onto glass. A common approach utilizes an array of fine pins or needles controlled by a robotic arm that is dipped into wells containing DNA probes and then depositing each probe at designated locations on the array surface. The resulting “grid” of probes represents the nucleic acid profiles of the prepared probes and is ready to receive complementary cDNA or cRNA “targets” derived from experimental or clinical samples. This technique is used by research scientists around the world to produce “in-house” printed microarrays from their own labs. These arrays may be easily customized for each experiment, because researchers can choose the probes and printing locations on the arrays, synthesize the probes in their own lab (or collaborating facility), and spot the arrays. They can then generate their own labeled samples for hybridization, hybridize the samples to the array, and finally scan the arrays with their own equipment. This provides a relatively low-cost microarray that may be customized for each study, and avoids the costs of purchasing often more expensive commercial arrays that may represent vast numbers of genes that are not of interest to the investigator. Publications exist which indicate in-house spotted microarrays may not provide the same level of sensitivity compared to commercial oligonucleotide arrays, possibly owing to the small batch sizes and reduced printing efficiencies when compared to industrial manufactures of oligo arrays.
In oligonucleotide microarrays, the probes are short sequences designed to match parts of the sequence of known or predicted open reading frames. Although oligonucleotide probes are often used in “spotted” microarrays, the term “oligonucleotide array” most often refers to a specific technique of manufacturing. Oligonucleotide arrays are produced by printing short oligonucleotide sequences designed to represent a single gene or family of gene splice-variants by synthesizing this sequence directly onto the array surface instead of depositing intact sequences. Sequences may be longer (60-mer probes such as the Agilent design) or shorter (25-mer probes produced by Affymetrix) depending on the desired purpose; longer probes are more specific to individual target genes, shorter probes may be spotted in higher density across the array and are cheaper to manufacture. One technique used to produce oligonucleotide arrays include photolithographic synthesis (Affymetrix) on a silica substrate where light and light-sensitive masking agents are used to “build” a sequence one nucleotide at a time across the entire array. Each applicable probe is selectively “unmasked” prior to bathing the array in a solution of a single nucleotide, then a masking reaction takes place and the next set of probes are unmasked in preparation for a different nucleotide exposure. After many repetitions, the sequences of every probe become fully constructed. More recently, Maskless Array Synthesis from NimbleGen Systems has combined flexibility with large numbers of probes.
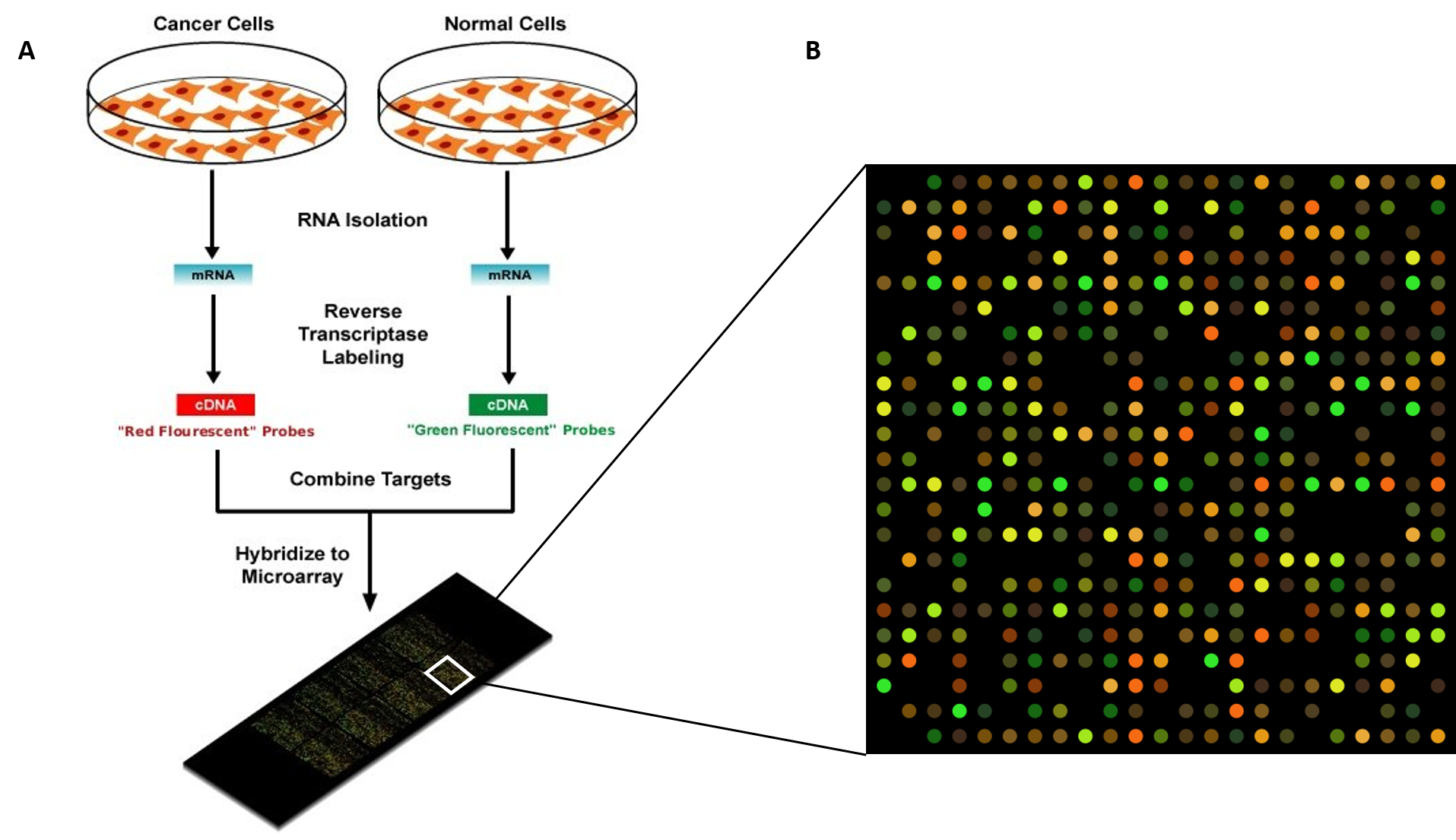
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
.jpg){kind=link}
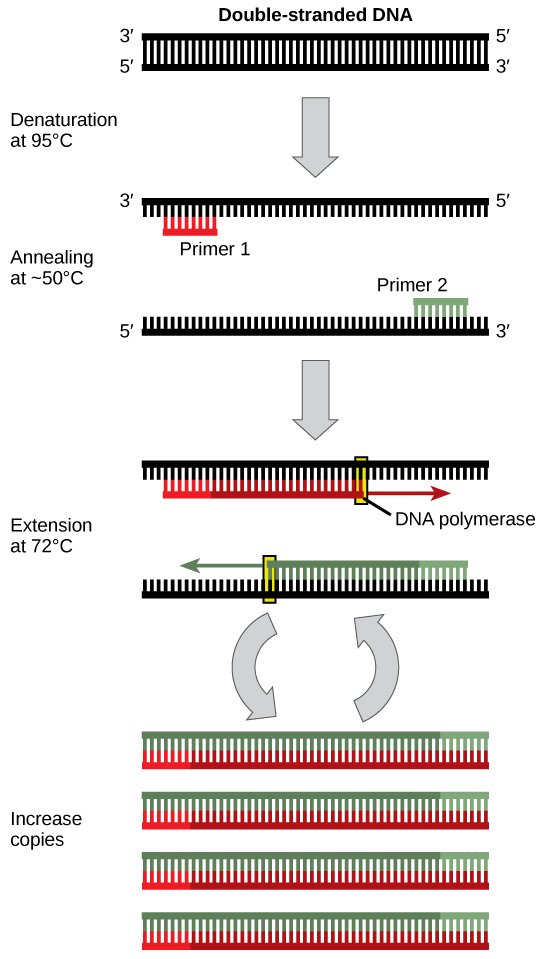{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5.27 Diagram of typical dual-color microarray experiment. Within a dual-color microarray, the probe DNA is typically hybridized with cDNA prepared from two different samples, each labelled with a different fluorescent probe. Analysis will yield green fluorescence for one sample that upregulates gene expression, whereas the other sample tagged with a red fluorescence marker will indicate that the other condition elicits gene expression at that location. Yellow indicates gene expression in both samples.
Image A modified from Larssono and Image B from Guillaume Paumier
{kind=link}
{kind=link}
Two-color microarrays or two-channel microarrays are typically hybridized with cDNA prepared from two samples to be compared (e.g. diseased tissue versus healthy tissue) and that are labeled with two different fluorophores. Fluorescent dyes commonly used for cDNA labeling include Cy3, which has a fluorescence emission wavelength of 570 nm (corresponding to the green part of the light spectrum), and Cy5 with a fluorescence emission wavelength of 670 nm (corresponding to the red part of the light spectrum). The two Cy-labeled cDNA samples are mixed and hybridized to a single microarray that is then scanned in a microarray scanner to visualize fluorescence of the two fluorophores after excitation with a laser beam of a defined wavelength. Relative intensities of each fluorophore may then be used in ratio-based analysis to identify up-regulated and down-regulated genes.
Oligonucleotide microarrays often carry control probes designed to hybridize with RNA spike-ins. The degree of hybridization between the spike-ins and the control probes is used to normalize the hybridization measurements for the target probes. Although absolute levels of gene expression may be determined in the two-color array in rare instances, the relative differences in expression among different spots within a sample and between samples is the preferred method of data analysis for the two-color system. Examples of providers for such microarrays includes Agilent with their Dual-Mode platform, Eppendorf with their DualChip platform for colorimetric Silverquant labeling, and TeleChem International with Arrayit.
In single-channel microarrays or one-color microarrays, the arrays provide intensity data for each probe or probe set indicating a relative level of hybridization with the labeled target. However, they do not truly indicate abundance levels of a gene but rather relative abundance when compared to other samples or conditions when processed in the same experiment. Each RNA molecule encounters protocol and batch-specific bias during amplification, labeling, and hybridization phases of the experiment making comparisons between genes for the same microarray uninformative. The comparison of two conditions for the same gene requires two separate single-dye hybridizations. Several popular single-channel systems are the Affymetrix “Gene Chip”, Illumina “Bead Chip”, Agilent single-channel arrays, the Applied Microarrays “CodeLink” arrays, and the Eppendorf “DualChip & Silverquant”. One strength of the single-dye system lies in the fact that an aberrant sample cannot affect the raw data derived from other samples, because each array chip is exposed to only one sample (as opposed to a two-color system in which a single low-quality sample may drastically impinge on overall data precision even if the other sample was of high quality). Another benefit is that data are more easily compared to arrays from different experiments as long as batch effects have been accounted for.
5.5 In Situ Hybridization
In situ hybridization (ISH) is a type of hybridization that uses a labeled complementary DNA, RNA or modified nucleic acids strand (i.e., probe) to localize a specific DNA or RNA sequence in a portion or section of tissue (in situ) or if the tissue is small enough (e.g., plant seeds, Drosophila embryos), in the entire tissue (whole mount ISH), in cells, and in circulating tumor cells (CTCs). This is distinct from immunohistochemistry, which usually localizes proteins in tissue sections.
In situ hybridization is used to reveal the location of specific nucleic acid sequences on chromosomes or in tissues, a crucial step for understanding the organization, regulation, and function of genes. The key techniques currently in use include in situ hybridization to mRNA with oligonucleotide and RNA probes (both radio-labeled and hapten-labeled), analysis with light and electron microscopes, whole mount in situ hybridization, double detection of RNAs and RNA plus protein, and fluorescent in situ hybridization to detect chromosomal sequences. DNA ISH can be used to determine the structure of chromosomes. Fluorescent DNA ISH (FISH) can, for example, be used in medical diagnostics to assess chromosomal integrity. RNA ISH (RNA in situ hybridization) is used to measure and localize RNAs (mRNAs, lncRNAs, and miRNAs) within tissue sections, cells, whole mounts, and circulating tumor cells (CTCs). In situ hybridization was invented by Mary-Lou Pardue and Joseph G. Gall.

Figure 5.28 In Situ Hybridization of wild type Drosophila embryos at different developmental stages for the RNA from a gene called hunchback.
Image by Nina
{kind=link}
For hybridization histochemistry, sample cells and tissues are usually treated to fix the target transcripts in place and to increase access of the probe. As noted above, the probe is either a labeled complementary DNA or, now most commonly, a complementary RNA (riboprobe). The probe hybridizes to the target sequence at elevated temperature, and then the excess probe is washed away (after prior hydrolysis using RNase in the case of unhybridized, excess RNA probe). Solution parameters such as temperature, salt, and/or detergent concentration can be manipulated to remove any non-identical interactions (i.e., only exact sequence matches will remain bound). Then, the probe that was labeled with either radio-, fluorescent- or antigen-labeled bases (e.g., digoxigenin) is localized and quantified in the tissue using either autoradiography, fluorescence microscopy, or immunohistochemistry, respectively. ISH can also use two or more probes, labeled with radioactivity or the other non-radioactive labels, to simultaneously detect two or more transcripts.
An alternative technology, branched DNA assay, can be used for RNA (mRNA, lncRNA, and miRNA ) in situ hybridization assays with single molecule sensitivity without the use of radioactivity. This approach (e.g., ViewRNA assays) can be used to visualize up to four targets in one assay, and it uses patented probe design and bDNA signal amplification to generate sensitive and specific signals. Samples (cells, tissues, and CTCs) are fixed, then treated to allow RNA target accessibility (RNA un-masking). Target-specific probes hybridize to each target RNA. Subsequent signal amplification is predicated on specific hybridization of adjacent probes (individual oligonucleotides [oligos] that bind side by side on RNA targets). A typical target-specific probe will contain 40 oligonucleotides, resulting in 20 oligo pairs that bind side-by-side on the target for detection of mRNA and lncRNA, and 2 oligos or a single pair for miRNA detection. Signal amplification is achieved via a series of sequential hybridization steps. A pre-amplifier molecule hybridizes to each oligo pair on the target-specific RNA, then multiple amplifier molecules hybridize to each pre-amplifier. Next, multiple label probe oligonucleotides (conjugated to alkaline phosphatase or directly to fluorophores) hybridize to each amplifier molecule. A fully assembled signal amplification structure “Tree” has 400 binding sites for the label probes. When all target-specific probes bind to the target mRNA transcript, an 8,000 fold signal amplification occurs for that one transcript. Separate but compatible signal amplification systems enable the multiplex assays. The signal can be visualized using a fluorescence or brightfield microscope.
5.6 References
EMBL-EBI (2012) Next generation sequencing practical course. Buplished by EMBL-EBI. Available at: https://www.ebi.ac.uk/training/online/course/ebi-next-generation-sequencing-practical-course/how-take-course
Ghannam, M.G., and Varacallo, M. (2018) Biochemistry, Polymerase Chain Reaction (PCR) StatPearls Publishing. Available at: https://www.ncbi.nlm.nih.gov/books/NBK535453/
Kahn, N.T. (2018) The Emerging Role of bioinformatics in Biotechnology. J. Biotech. and Biomed. Science 1(3) ISSN: 2576-6694. Available at: https://openaccesspub.org/jbbs/article/803
Lee LYY, Izzard L and Hurt AC (2018) A Review of DNA Vaccines Against Influenza. Front. Immunol. 9:1568. doi: 10.3389/fimmu.2018.01568 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6046547/pdf/fimmu-09-01568.pdf
Molnar, C. and Gair, J. (2019) 10.1 Cloning and Genetic Engineering. Chapter in Concepts of Biology – 1st Candadian Edition. Available at: https://opentextbc.ca/biology/
Nidhi M. Bioinformatics: An Introduction. Open Acc Biostat Bioinform. 1(4). OABB.000522. 2018. DOI: 10.31031/OABB.2018.01.000522 https://pdfs.semanticscholar.org/f220/86467e2532106c8c616f03fc0a61aff9b3ea.pdf
Seto, D. (2010) Viral genomics and Bioinformatics. Viruses 2: 2587-2593. doi:10.3390/v2122587
Seto, J. (2019) Restriction Enzymes. Chapter in Biology OER, published by The OpenLab at City Tech. Available at: https://openlab.citytech.cuny.edu/bio-oer/analyzing-dna/restriction-enzymes/
Wikipedia contributors. (2019, July 8). Complementary DNA. In Wikipedia, The Free Encyclopedia. Retrieved 15:18, September 8, 2019, from https://en.wikipedia.org/w/index.php?title=Complementary_DNA&oldid=905281015
Wikipedia contributors. (2019, May 15). Genomic DNA. In Wikipedia, The Free Encyclopedia. Retrieved 15:25, September 8, 2019, from https://en.wikipedia.org/w/index.php?title=Genomic_DNA&oldid=897250028
Wikipedia contributors. (2019, July 10). DNA extraction. In Wikipedia, The Free Encyclopedia. Retrieved 05:05, September 9, 2019, from https://en.wikipedia.org/w/index.php?title=DNA_extraction&oldid=905644602
Wikipedia contributors. (2018, May 9). Spin column-based nucleic acid purification. In Wikipedia, The Free Encyclopedia. Retrieved 05:26, September 9, 2019, from https://en.wikipedia.org/w/index.php?title=Spin_column-based_nucleic_acid_purification&oldid=840437141
Wikipedia contributors. (2019, August 29). DNA sequencing. In Wikipedia, The Free Encyclopedia. Retrieved 05:31, September 9, 2019, from https://en.wikipedia.org/w/index.php?title=DNA_sequencing&oldid=913023155
Wikipedia contributors. (2019, July 30). Sanger sequencing. In Wikipedia, The Free Encyclopedia. Retrieved 05:28, September 10, 2019, from https://en.wikipedia.org/w/index.php?title=Sanger_sequencing&oldid=908599177
Wikipedia contributors. (2019, June 5). Artificial gene synthesis. In Wikipedia, The Free Encyclopedia. Retrieved 19:19, September 13, 2019, from https://en.wikipedia.org/w/index.php?title=Artificial_gene_synthesis&oldid=900437337
Wikipedia contributors. (2019, September 16). Cloning vector. In Wikipedia, The Free Encyclopedia. Retrieved 18:26, September 26, 2019, from https://en.wikipedia.org/w/index.php?title=Cloning_vector&oldid=916010157
Wikipedia contributors. (2019, October 2). CRISPR. In Wikipedia, The Free Encyclopedia. Retrieved 04:12, October 8, 2019, from https://en.wikipedia.org/w/index.php?title=CRISPR&oldid=919232229
Wikipedia contributors. (2019, September 28). DNA microarray. In Wikipedia, The Free Encyclopedia. Retrieved 04:53, October 8, 2019, from https://en.wikipedia.org/w/index.php?title=DNA_microarray&oldid=918427523
Wikipedia contributors. (2019, September 22). In situ hybridization. In Wikipedia, The Free Encyclopedia. Retrieved 04:59, October 8, 2019, from https://en.wikipedia.org/w/index.php?title=In_situ_hybridization&oldid=917168202