Home » Student Resources » Online Chemistry Textbooks » CH450 and CH451: Biochemistry - Defining Life at the Molecular Level » Chapter 2: Protein Structure
MenuCH450 and CH451: Biochemistry - Defining Life at the Molecular Level
Chapter 2: Protein Structure
2.1 Amino Acid Structure and Properties
2.2 Peptide Bond Formation and Primary Protein Structure
2.3 Secondary Protein Structure
2.4 Supersecondary Structure and Protein Motifs
2.5 Tertiary and Quaternary Protein Structure
2.6 Protein Folding, Denaturation and Hydrolysis
2.7 References
2.1 Amino Acid Structure and Properties
Proteins are one of the most abundant organic molecules in living systems and have the most diverse range of functions of all macromolecules. Proteins may be structural, regulatory, contractile, or protective; they may serve in transport, storage, or membranes; or they may be toxins or enzymes. Each cell in a living system may contain thousands of different proteins, each with a unique function. Their structures, like their functions, vary greatly. They are all, however, polymers of alpha amino acids, arranged in a linear sequence and connected together by covalent bonds.
Alpha Amino Acid Structure
The major building block of proteins are called alpha (α) amino acids. As their name implies they contain a carboxylic acid functional group and an amine functional group. The alpha designation is used to indicate that these two functional groups are separated from one another by one carbon group. In addition to the amine and the carboxylic acid, the alpha carbon is also attached to a hydrogen and one additional group that can vary in size and length. In the diagram below, this group is designated as an R-group. Within living organisms there are 20 amino acids used as protein building blocks. They differ from one another only at the R-group position. The basic structure of an amino acid is shown below:
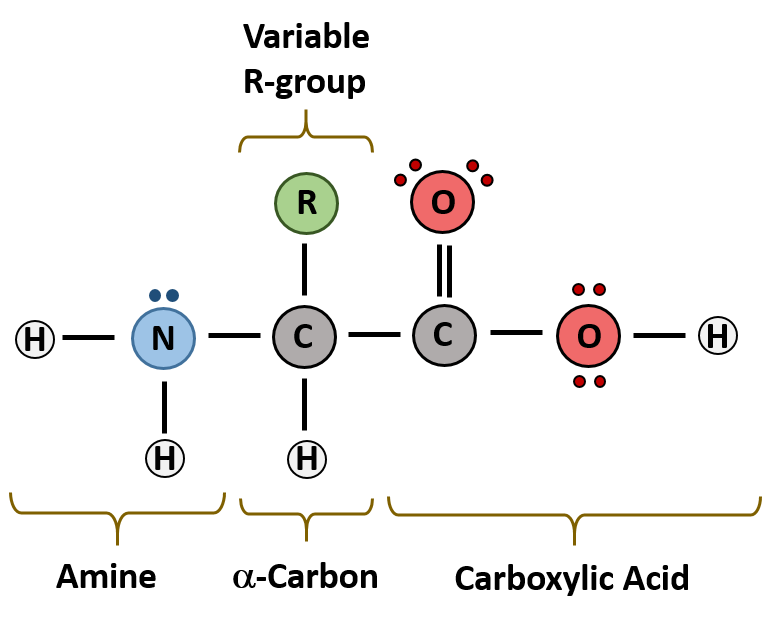
Figure 2.1 General Structure of an Alpha Amino Acid
There are a total of 20 alpha amino acids that are commonly incorporated into protein structures (Figure 2.x). The different R-groups have different characteristics based on the nature of atoms incorporated into the functional groups. There are R-groups that predominantly contain carbon and hydrogen and are very nonpolar or hydrophobic. Others contain polar uncharged functional groups such as alcohols, amides, and thiols. A few amino acids are basic (containing amine functional groups) or acidic (containing carboxylic acid functional groups). These amino acids are capable of forming full charges and can have ionic interactions. Each amino acid can be abbreviated using a three letter and a one letter code.
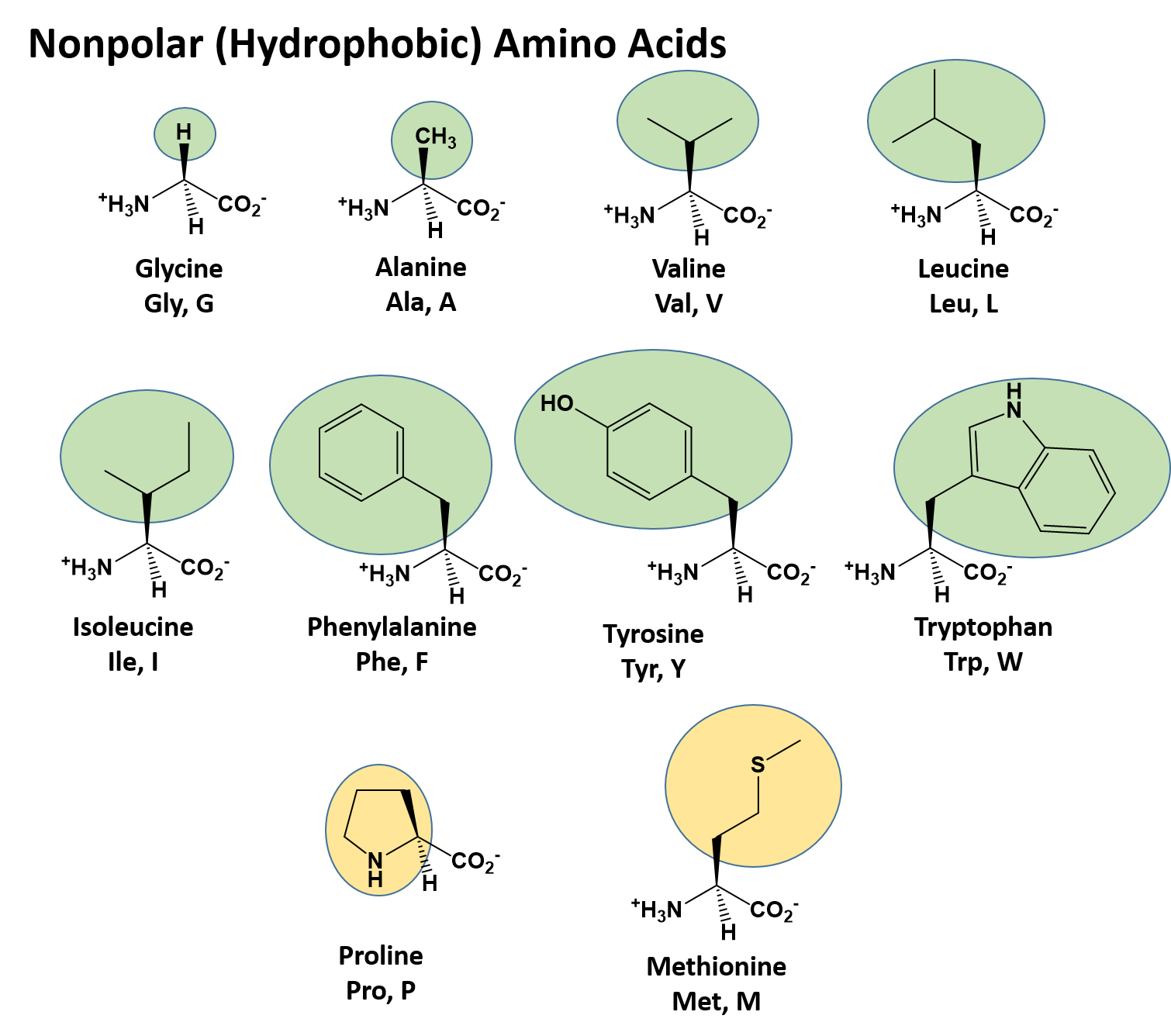
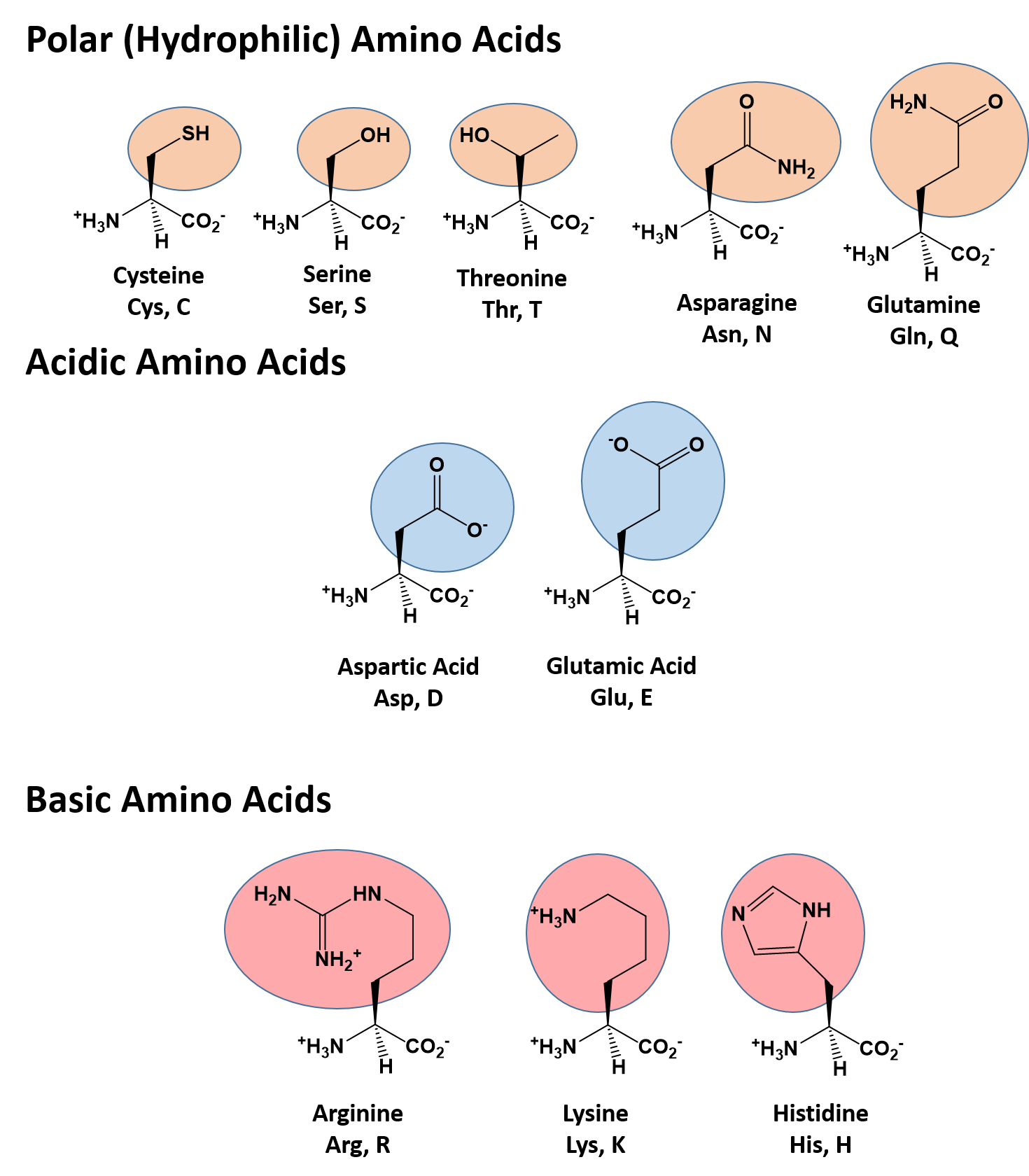
Figure 2.2 Structure of the 20 Alpha Amino Acids used in Protein Synthesis. R-groups are indicated by circled/colored portion of each molecule. Colors indicate specific amino acid classes: Hydrophobic – Green and Yellow, Hydrophilic Polar Uncharged – Orange, Hydrophilic Acidic – Blue, Hydrophilic Basic – Rose.
Click Here for a Downloadable Version of the Amino Acid Chart
Nonpolar (Hydrophobic) Amino Acids
The nonpolar amino acids can largely be subdivided into two more specific classes, the aliphatic amino acids and the aromatic amino acids. The aliphatic amino acids (glycine, alanine, valine, leucine, isoleucine, and proline) typically contain branched hydrocarbon chains with the simplest being glycine to the more complicated structures of leucine and valine. Proline is also classified as an aliphatic amino acid but contains special properties as the hydrocarbon chain has cyclized with the terminal amine creating a unique 5-membered ring structure. As we will see in the next section covering primary structure, proline can significantly alter the 3-dimentional structure of the due to the structural rigidity of the ring structure when it is incorporated into the polypeptide chain and is commonly found in regions of the protein where folds or turns occur.
The aromatic amino acids (phenylalanine, tyrosine, and tryptophan), as their name implies, contain an aromatic functional groups within their structure making them largely nonpolar and hydrophobic due to the high carbon/hydrogen content. However, it should be noted that hydrophobicity and hydrophilicity represent a sliding scale and each of the different amino acids can have different physical and chemical properties depending on their structure. For example, the hydroxyl group present in tyrosine increase its reactivity and solubility compared to that of phenylalanine.
Methionine, one of the sulfur-containing amino acids is usually classified under the nonpolar, hydrophobic amino acids as the terminal methyl group creates a thioether functional group which generally cannot form a permanent dipole within the molecule and retains low solubility.
Polar (Hydrophilic) Amino Acids
The polar, hydrophilic amino acids can be subdivided into three major classes, the polar uncharged-, the acidic-, and the basic- functional groups. Within the polar uncharged class, the side chains contain heteroatoms (O, S, or N) that are capable of forming permanent dipoles within the R-group. These include the hydroxyl- and sulfoxyl-containing amino acids, serine, threonine, and cysteine, and the amide-containing amino acids, glutamine and asparigine. Two amino acids, glutamic acid (glutamate), and aspartic acid (aspartate) constitute the acidic amino acids and contain side chains with carboxylic acid functional groups capable of fully ionizing in solution. The basic amino acids, lysine, arginine, and histidine contain amine functional groups that can be protonated to carry a full charge.
Many of the amino acids with hydrophilic R-groups can participate within the active site of enzymes. An active site is the part of an enzyme that directly binds to a substrate and carries a reaction. Protein-derived enzymes contain catalytic groups consisting of amino acid R-groups that promote formation and degradation of bonds. The amino acids that play a significant role in the binding specificity of the active site are usually not adjacent to each other in the primary structure, but form the active site as a result of folding in creating the tertiary structure, as you will see later in the chapter.
Protein structures built from the basic amino acids can be hundreds of amino acids long. Thus, for simplicity sake, the 20 amino acids used for protein synthesis have both three letter and one letter code abbreviations (Table 2.1). These abbreviations are commonly used to delineate protein sequences for bioinformatic and research purposes.
Table 2.1 α-Amino Acid Abbreviations

Thought Question: Tryptophan contains an amine functional group, why isn’t tryptophan basic?
Answer: Tryptophan contains an indole ring structure that includes the amine functional group. However, due to the proximity of, and electron withdrawing nature of the aromatic ring structure, the lone pair of electrons on the nitrogen are unavailable to accept a proton. Instead they are involved in forming pi-bonds within several of the different resonance structures possible for the indole ring. Figure 2.3A shows four of the possible resonance structures for indole. Conversely, within the immidazole ring structure found in histidine, there are two nitrogen atoms, one of which is involved in the formation of resonance structures (Nitrogen #1 in Figure 2.3B) and cannot accept a proton, and the other (Nitrogen #3) that has a lone pair of electrons that is available to accept a proton.

Figure 2.3 Comparison of the Structural Availability of Lone Pair of Electrons on Nitrogen to Accept a Proton in the Indole and Immidizole Ring Structures. (A) Shown are four resonance structures of the indole ring structure demonstrating that the lone pair of electrons on the nitrogen are involved in the formation of pi-bonds. (B) The immidazole ring structure has one nitrogen (1) that is involved in resonance structures (not shown) and is not available to accept a proton, while the second nitrogen (3) has a lone pair of electrons available to accept a proton as shown.
Work It Out on Your Own:
Given the example above, describe using a chemical diagram, why the amide nitrogen atoms found in asparagine and glutamine are not basic.
Alpha Amino Acids are Chiral Molecules
If you examine the structure of the alpha carbon within each of the amino acids, you will notice that all of the amino acids, except for glycine are chiral molecules (Figure 2.4) A chiral molecule is one that is not superimposable with its mirror image. Like left and right hands that have a thumb, fingers in the same order, but are mirror images and not the same, chiral molecules have the same things attached in the same order, but are mirror images and not the same. The mirror image versions of chiral molecules have physical properties that are nearly identical to one another, making it very difficult to tell them apart from one another or to separate. Because of this nature, they are given a special stereoisomer name called enantiomers and in fact, the compounds themselves are given the same name! These molecules do differ in the way that they rotate plain polarized light and the way that they react with and interact with biological molecules. Molecules that rotate the light in the right-handed direction are called dextrorotary and are given a D- letter designation. Molecules that rotate light in the left-handed direction are called levorotary and are give an L- letter designation to distinguish one enantiomer from the other. The D- and L- forms of alanine are show in Figure 2.4B.
Although most amino acids can exist in both left and right handed forms, life on Earth is made of left handed amino acids, almost exclusively. Proteogenic amino acids incorporated into proteins by ribosomes are always in the L-conformation. Some bacteria can incorporate D-amino acids into non-ribosomally encoded peptides, but the use of D-amino acids in nature is rare. Interestingly, when we will discuss the structure of sugars in Chapter XX, we will find that sugars that are incorporated into carbohydrate structures are almost exclusively in the D-conformation. No one knows why this is the case. However, Drs. John Cronin and Sandra Pizzarello have shown that of the amino acids that fall to earth from space on meteorites, more are in the L-conformation than the D-conformation. Thus, the fact that we are made predominantly of L-amino acids may be because of amino acids from space.
Why do amino acids in space favor the L-conformation? No one really knows, but it is known that radiation can also exist in left and right handed forms. So, there is a theory called the Bonner hypothesis, that proposes that the predominant forms of radiation in space (ie. from a rotating neutron star for example) could lead to the selective formation of homochiral molecules, such as L-amino acids and D-sugars. This is still speculative, but recent findings from meteorites make this hypothesis much more plausible.
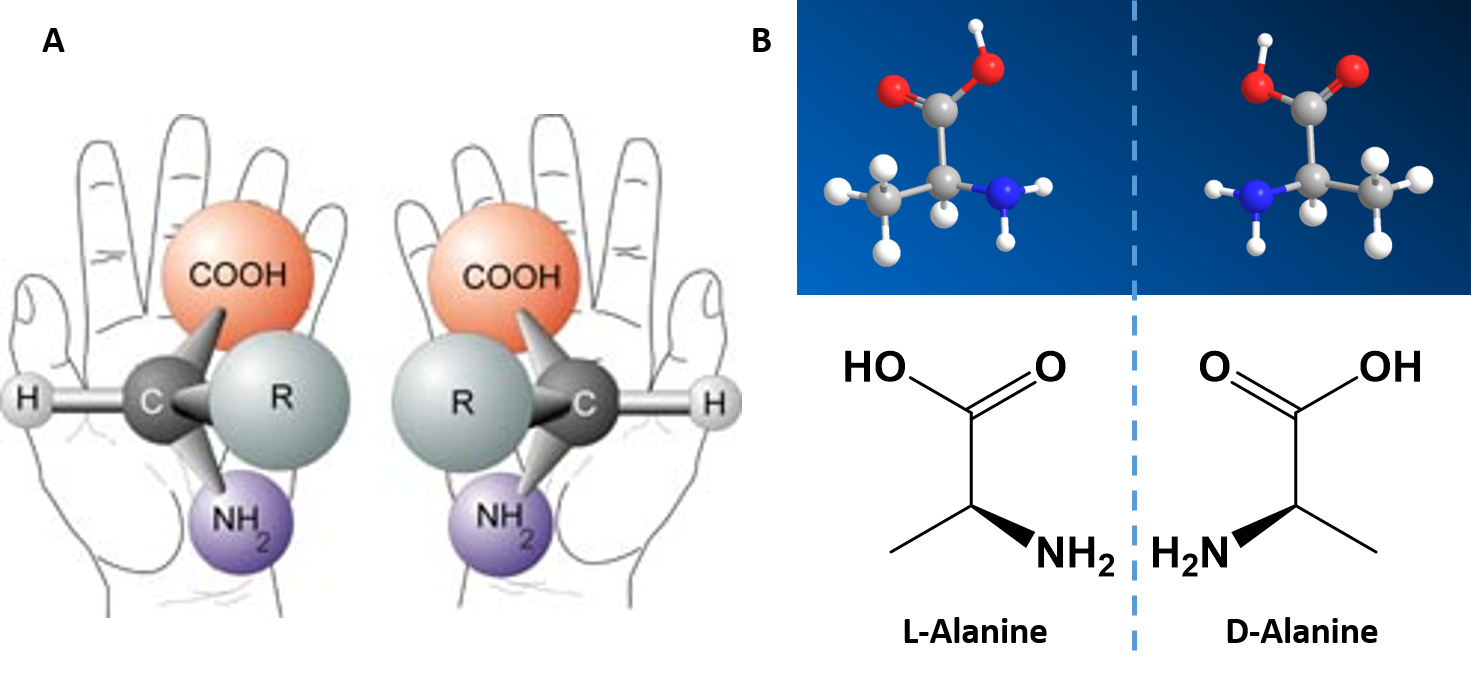
Figure 2.4 Amino Acid Chirality. Except for the simplest amino acid, glycine, all of the other amino acids that are incorporated into protein structures are chiral in nature. (A) Demonstrates the chirality of the core alpha amino acid structure when the non-specific R-group is used. (B) The D- and L-Alanine enantiomer pair, upper diagram represents the ball and stick model and the lower diagram represents the line structure.
Image (A) from NASA
Note that the D- and L-designations are specific terms used for the way a molecule rotates plain polarized light. It does not denote the absolute stereo configuration of a molecule. An absolute configuration refers to the spatial arrangement of the atoms of a chiral molecular entity (or group) and its stereochemical description e.g. R or S, referring to Rectus, or Sinister, respectively.
Absolute configurations for a chiral molecule (in pure form) are most often obtained by X-ray crystallography. Alternative techniques are optical rotatory dispersion, vibrational circular dichroism, use of chiral shift reagents in proton NMR and Coulomb explosion imaging. When the absolute configuration is obtained the assignment of R or S is based on the Cahn–Ingold–Prelog priority rules, which can be reviewed by following the link and in Figure 2.5. All of the chiral amino acids, except for cysteine, are also in the S-conformation. Cysteine, contains the sulfur atom causing the R-group to have higher priority than the carboxylic acid functional group, leading to the R-conformation for the absolute stereochemistry. However, cysteine does rotate plain polarized light in the levorotary or left-handed direction. Thus, the R- and S-designations do not always correspond with the D- and L- conformation.
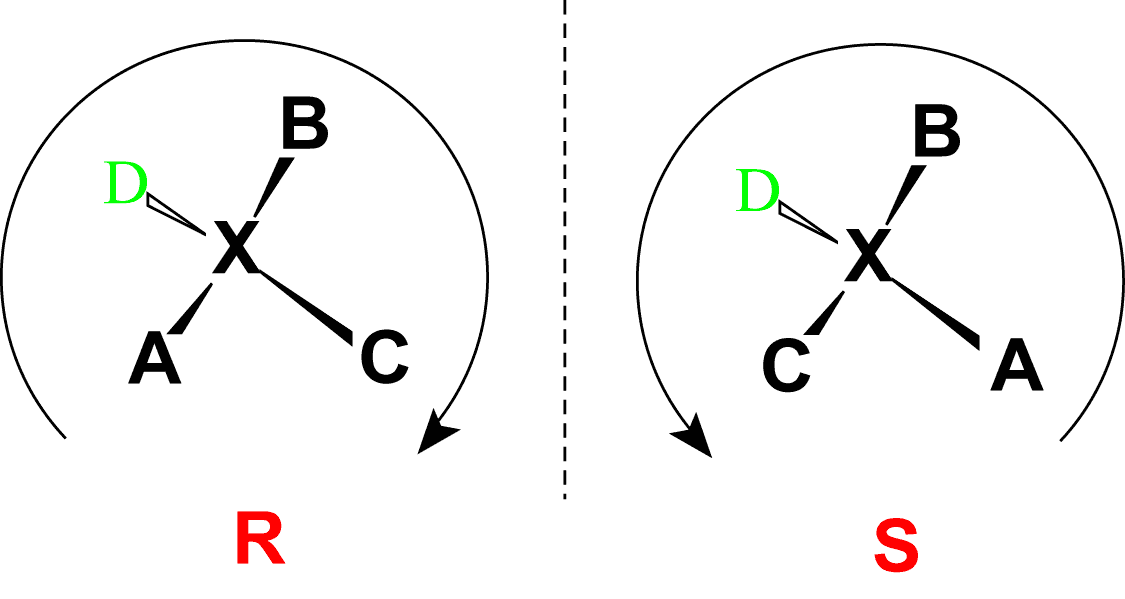
Figure 2.5 Absolute Configuration is Determined by the Rectus (R) and Sinister (S) Designations. In the Cahn Ingold Prelog system for naming chiral centers, the groups attached to the chiral center are ranked according to their atomic number with the highest atomic number receiving the highest priority (A in the diagram above) and the lowest atomic number receiving the lowest priority (D in the diagram above). The lowest priority is then pointed away from the viewer to correctly orient the molecule for further evaluation. The path of priorities #1, #2, and #3 (corresponding to A, B and C above) are then traced. If the path is is in the clockwise direction, the chiral center is given the R-designation, whereas if the path is counterclockwise, it is given the S-designation.
Image from Wikipedia
Amino Acids are Zwitterions
In chemistry, a zwitterion is a molecule with two or more functional groups, of which at least one has a positive and one has a negative electrical charge and the net charge of the entire molecule is zero at a specific pH. Because they contain at least one positive and one negative charge, zwitterions are also sometimes called inner salts. The charges on the different functional groups balance each other out, and the molecule as a whole can be electrically neutral at a specific pH. The pH where this happens is known as the isoelectric point.
Unlike simple amphoteric compounds that may only form either a cationic or anionic species, a zwitterion simultaneously has both ionic states. Amino acids are examples of zwitterions (Figure 2.6). These compounds contain an ammonium and a carboxylate group, and can be viewed as arising via a kind of intramolecular acid–base reaction: The amine group deprotonates the carboxylic acid.

Figure 2.6 Amino Acids are Zwitterions. An amino acid contains both acidic (carboxylic acid fragment) and basic (amine fragment) centres. The isomer on the right is the zwitterionic form.
Because amino acids are zwitterions, and several also contain the potential for ionization within their R-groups, their charge state in vivo, and thus, their reactivity can vary depending on the pH, temperature, and solvation status of the local microenvironment in which they are located. The chart of standard pKa values for the amino acids is shown in Table 2.1 and can be used to predict the ionization/charge status of amino acids and their resulting peptides/proteins. However, it should be noted that the solvation status in the microenvironment of an amino acid can alter the relative pKa values of these functional groups and provide unique reactive properties within the active sites of enzymes (Table 2.1). A more in depth discussion of the effects of desolvation will be given in Chapter XX discussing enzyme reaction mechanisms.
Table 2.1
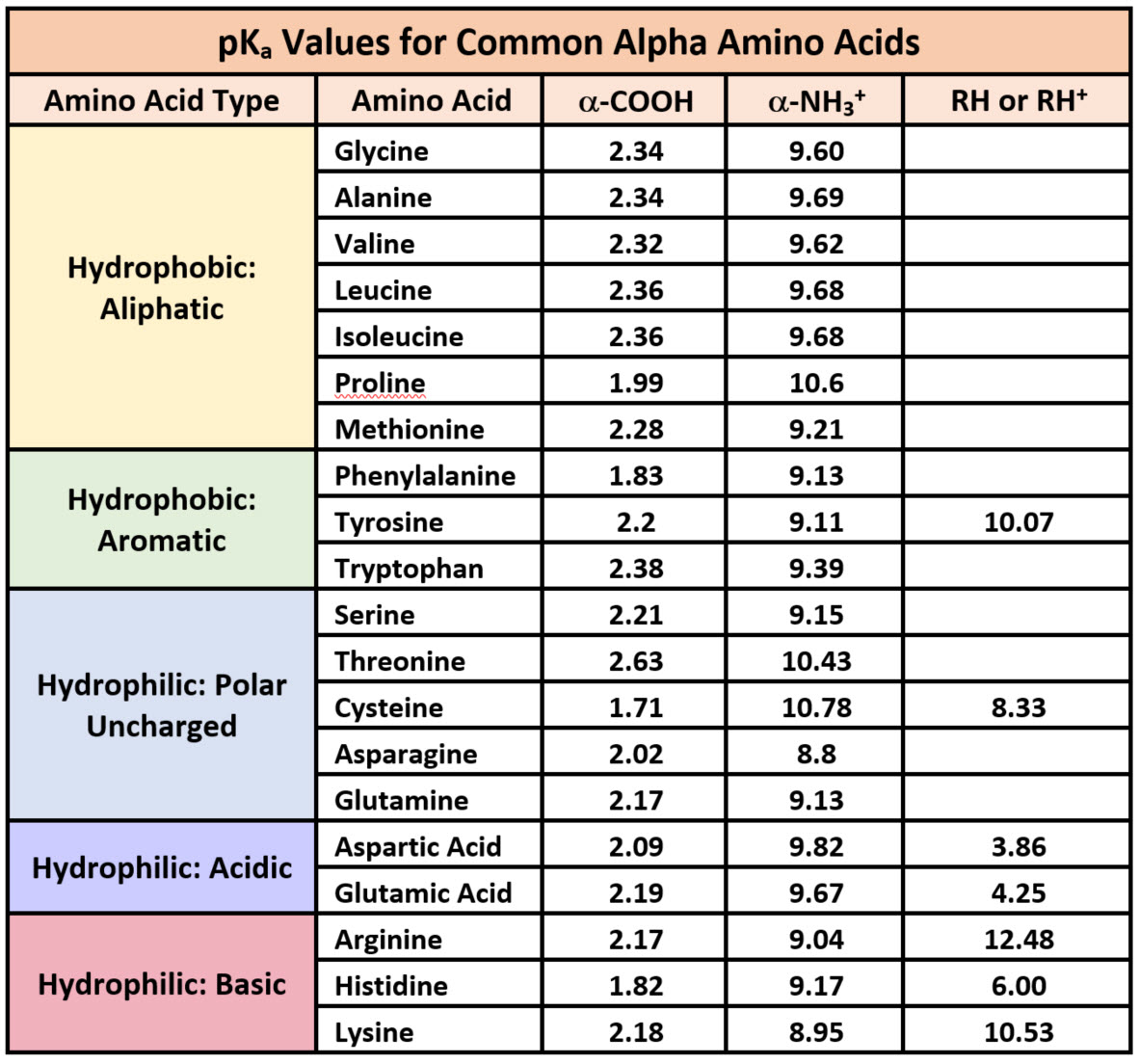
Printable Version of pKa Values
As seen in Table 2.1, seven of the amino acids contain R-groups with ionizable side chains and are commonly found in the active sites of enzymes. Recall that the pKa is defined as the pH at which the ionized and unionized forms of an ionizable functional group within a molecule exist in equal concentrations. Thus, as a functional group shifts above or below its pKa value, there will be a shift in the concentrations of the ionized and unionized forms favoring one state over the other. Figure 2.7 shows the various R-groups in their unionized and ionized states and their favored states either above or below the pKa value.
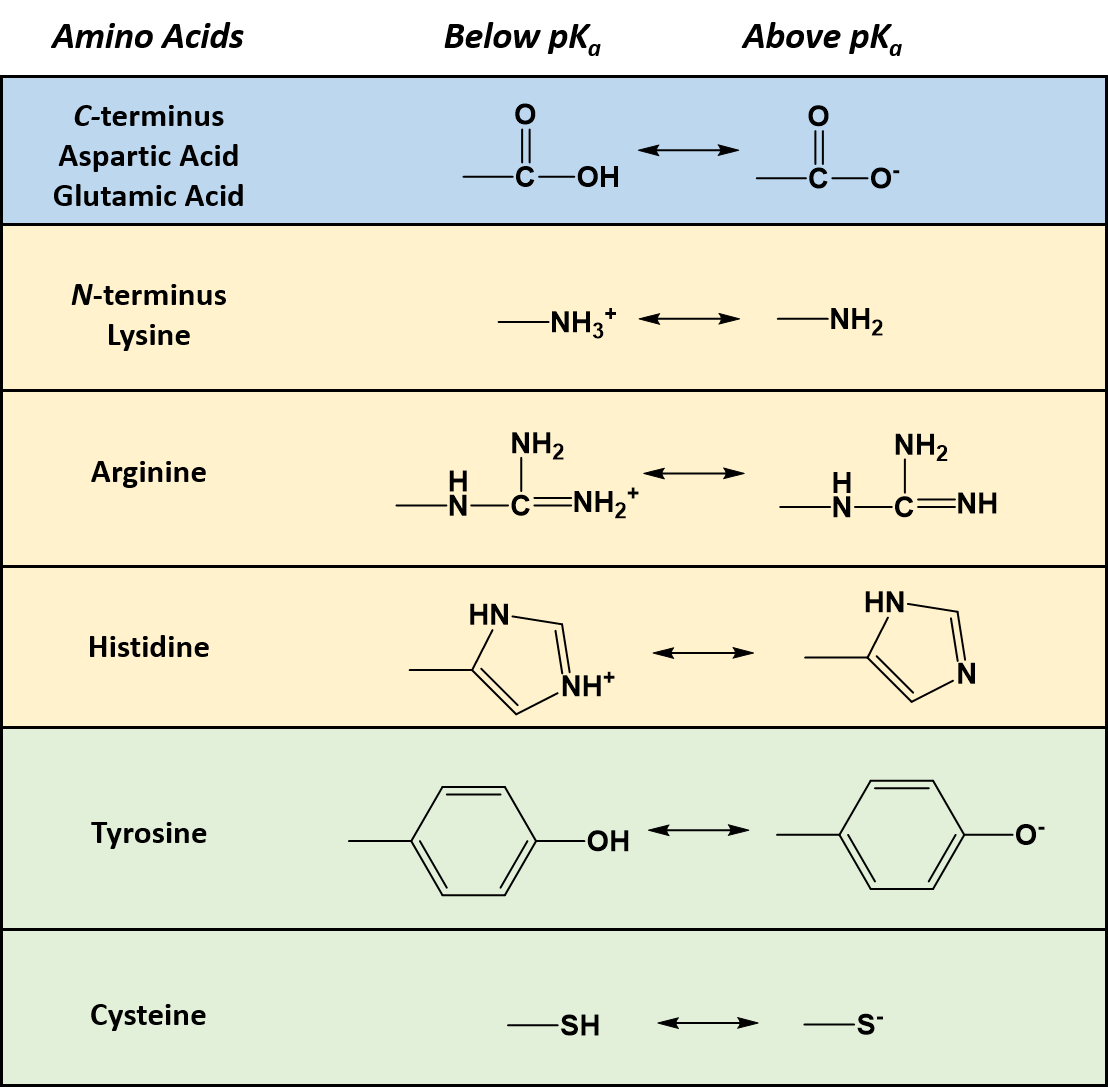
Figure 2.7 Ionizable Functional Groups in Common Amino Acids. Within all amino acids both the carboxylic acid functional group (C-terminus), and the amine functional group (N-terminus) are capable of ionization. In addition, seven amino acids (aspartic acid, glutamic acid, arginine, histidine, lysine, tyrosine, and cysteine) also contain ionizable functional groups within their R-groups. The functional group’s favored states are shown either above or below their respective pKa values.
Typically an ionizable group will favor the protonated state in pH conditions below its respective pKa values and will favor the deprotonated state in pH conditions above its respective pKa value. Thus, pKa values can be used to help predict the overall charge states of amino acids and their resulting peptides/proteins within a defined environment. For example, if we look at a titration curve for the basic amino acid, histidine (Figure 2.8). As each pKa is reached, the charge state of the amino acid is altered to favor the deprotonated state. Thus, histidine will slowly progress from an overall +2 charge at very low pH (fully protonated) to an overall -1 charge at very high pH (fully deprotonated).
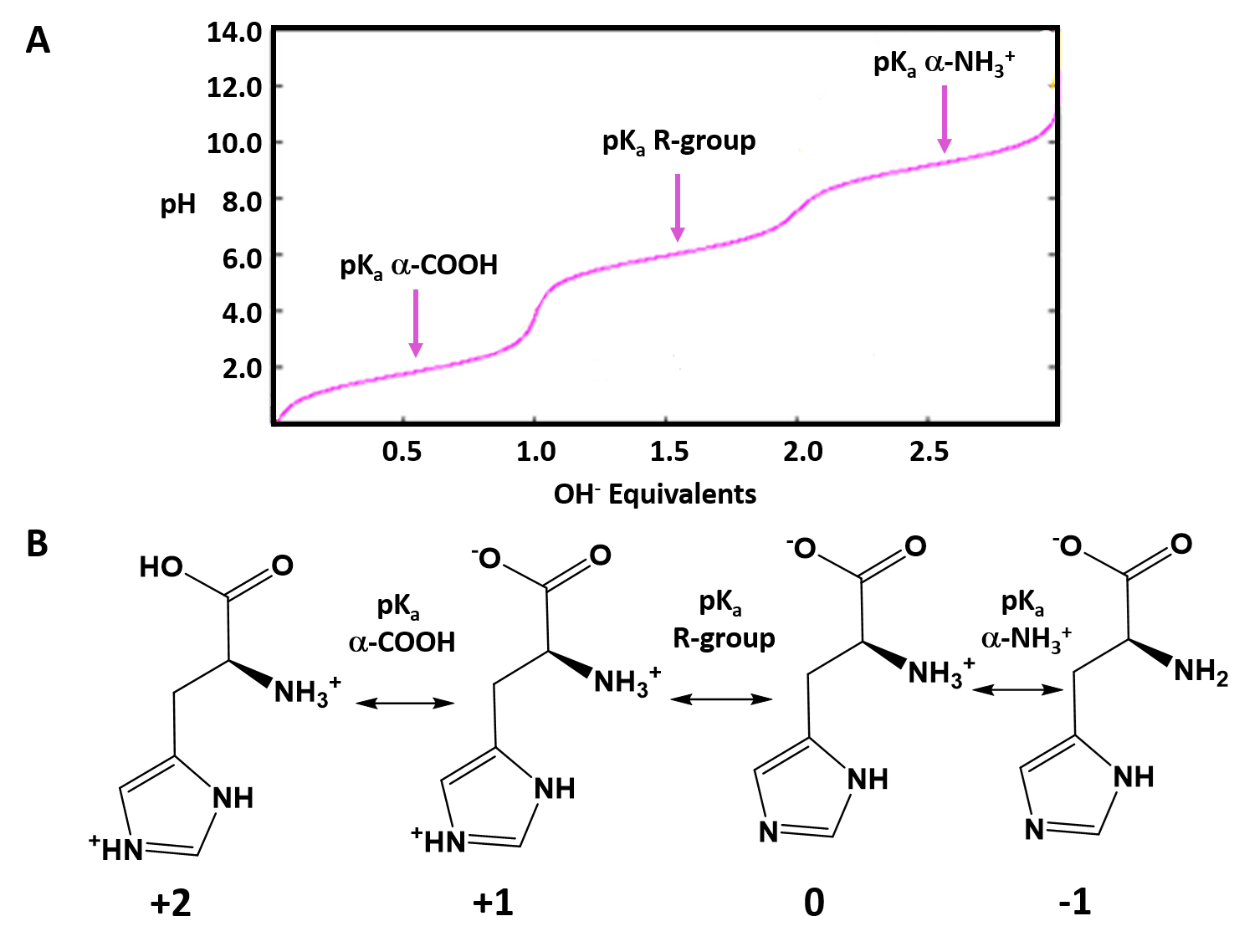
Figure 2.8 Ionization State of Histidine in Different pH Environments. (A) Titration curve of histidine from low pH to high pH. Each equivalence point (pKa) is indicated. (B) Shows the favored ionization state of histidine following the passage of each pKa value.
Image adapted from L. Van Warren
Extra Practice:
Draw glutamic acid and predict the overall charge state of the amino acid at pH = 1, pH = 3, pH = 7, and pH = 12.
Cysteine and Disulfide Bond Formation
Cysteine is also a unique amino acid as this side chain is capable of undergoing a reversible oxidation-reduction (redox) reaction with other cysteine residues creating a covalent disulfide bond in the oxidized state (Figure 2.9). Recall that when molecules become oxidized that they are losing electrons and that when molecules are reduced that they are gaining electrons. During biological redox reactions, hydrogen ions (protons) are often removed with the electrons from the molecule during oxidation, and are returned during reduction. Thus, if a reaction is losing or gaining protons, this is a good indication that it is also losing or gaining electrons and that a redox reaction is occurring. Thus, proton gain or loss can be an easy way to identify this reaction type.
Disulfide bonds are integral in the formation of the 3-dimentional structure of proteins and can therefore highly impact the function of the resulting protein. In cellular systems, disulfide bond formation/disruption is an enzyme-mediated reaction and can be utilized as a mechanism to control the activity of protein. Disulfide bonds will be discussed in further detail section 2.xx within this chapter and in Chapter XX.

Figure 2.9 Cysteine can be Oxidized to Produce Disulfide Bonds. During disulfide bond formation, two cysteines are oxidized to form a cystine molecule. This requires the loss of two protons and two electrons.
back to the top
2.2 Peptide Bond Formation and Primary Protein Structure
Within cellular systems, proteins are linked together by a large enzyme complex that contains a mixture of RNA and proteins. This complex is called the ribosome. Thus, as the amino acids are linked together to form a specific protein, they are placed within a very specific order that is dictated by the genetic information contained within the messenger RNA (mRNA) molecule. This specific ordering of amino acids is known as the protein’s primary sequence. The translation mechanism used by the ribosome to synthesize proteins will be discussed in detail in Chapter XX. This chapter will focus only on the chemical reaction occurring during synthesis and the physical properties of the resulting peptides/proteins.
The primary sequence of a protein is linked together using dehydration synthesis (loss of water) that combine the carboxylic acid of the upstream amino acid with the amine functional group of the downstream amino acid to form an amide linkage (Figure 2.10). Similarly, the reverse reaction is hydrolysis and requires the incorporation of a water molecule to separate two amino acids and break the amide bond. Notably, the ribosome serves as the enzyme that mediates the dehydration synthesis reactions required to build protein molecules, whereas a class of enzymes called proteases are required for protein hydrolysis.
Within protein structures, the amide linkage between amino acids is known as the peptide bond. Subsequent amino acids will be added onto the carboxylic acid terminal of the growing protein. Thus, proteins are always synthesized in a directional manner starting with the amine and ending with the carboxylic acid tail. New amino acids are always added onto the carboxylic acid tail, never onto the amine of the first amino acid in the chain. The directionality of protein synthesis is dictated by the ribosome and is known as N- to C- synthesis.
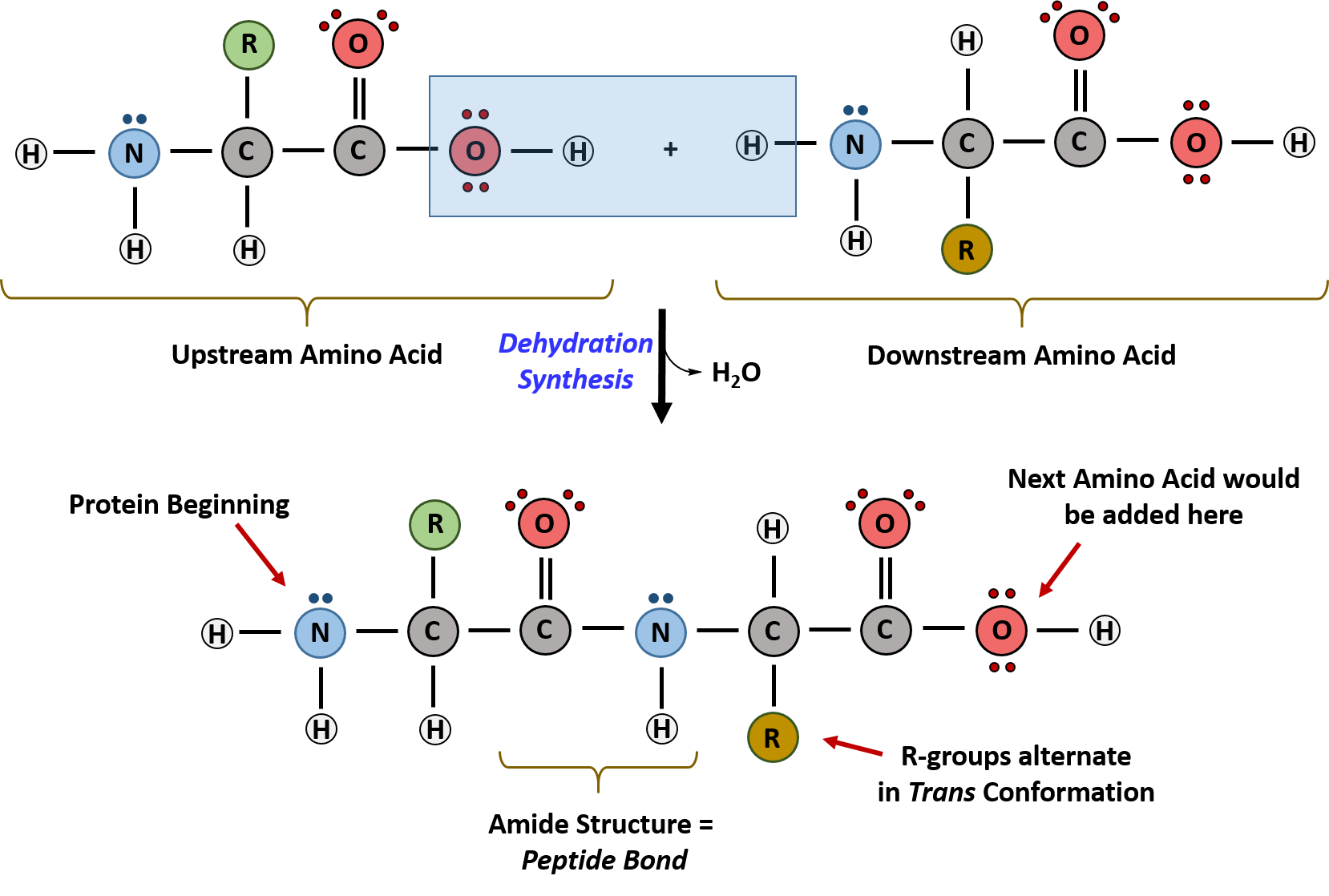
Figure 2.10 Formation of the Peptide Bond. The addition of two amino acids to form a peptide requires dehydration synthesis.
As noted above in the zwitterion section, amide bonds have a resonance structure that will not allow the nitrogen lone pair of electrons to act as a base (Figure 2.11).
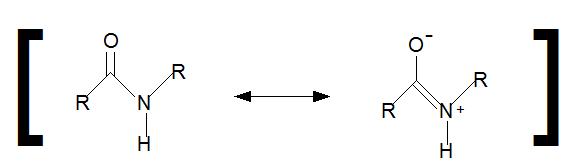
Figure 2.11 Amide Resonance Structure. During amide resonance, the lone pair electrons from the nitrogen are involved in pi-bond formation with the carbonyl carbon forming the double bond. Thus, amide nitrogens are not basic. In addition, the C-N bond within the amide structure is fixed in space and cannot rotate due to the pi-bond character.
Image from V.K. Chang
Instead, they are involved in pi-bond formation with the carbonyl carbon. Furthermore, the C-N bond within the amide structure is fixed in space and cannot rotate due to the pi-bond character. This creates fixed physical locations of the R-groups within the growing peptide in either the cis or trans conformations. Because the R-groups can be quite bulky, they usually alternate on either side of the growing protein chain in the trans conformation. The cis conformation is only preferred with one specific amino acid, proline. This is due to the cyclic structure of the proline R-group and the steric hindrance that is created when proline adopts the trans conformation (Figure 2.12). Thus, proline residues can have a large impact on the 3-D structure of the resulting peptide.
![]()
Figure 2.12 Cis and Trans Conformation of Amino Acid R-Groups. The upper diagram displays the cis and trans conformations of two adjacent amino acids noted as X and Y which indicate any of the 20 amino acids, except for proline. In the trans conformation the R-group from amino acid X is rotated away and on the other side of the molecule when compared with the R-group from amino acid Y. This conformation gives the least amount of steric hindrance compared with the cis conformation where the R-groups are located on the same side and in close proximity to one another. In the lower diagram, any amino acid, X is positioned upstream of a proline residue. Due to the cyclization of the proline R-group with the amide nitrogen in the backbone, this shifts the position of the proline R-group to be in closer proximity to the R-group from amino acid X when it adopts the trans conformation. Thus, proline favors the cis conformation which has less steric hindrance.
Proteins are very large molecules containing many amino acid residues linked together in very specific order. Proteins range in size from 50 amino acids in length to the largest known protein containing 33,423 amino acids. Macromolecules with fewer than 50 amino acids are known as peptides (Figure 2.13).
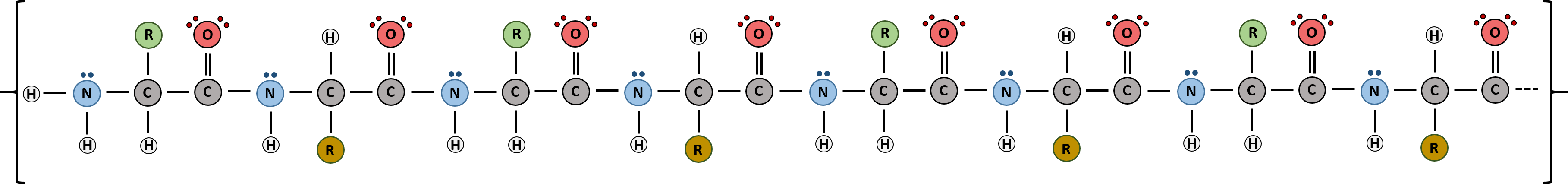
Figure 2.13 Peptides and Proteins are macromolecules built from long chains of amino acids joined together through amide linkages. The order and nature of amino acids in the primary sequence of a protein determine the folding pattern of the protein based on the surrounding environment of the protein (ie if it is inside the cell, it is likely surrounded by water in a very polar environment, whereas if the protein is embedded in the plasma membrane, it will be surrounded by very nonpolar hydrocarbon tails).
Due to the large pool of amino acids that can be incorporated at each position within the protein, there are billions of different possible protein combinations that can be used to create novel protein structures! For example, think about a tripeptide made from this amino acid pool. At each position there are 20 different options that can be incorporated. Thus, the total number of resulting tripeptides possible would be 20 X 20 X 20 or 203, which equals 8,000 different tripeptide options! Now think about how many options there would be for a small peptide containing 40 amino acids. There would be 2040 options, or a mind boggling 1.09 X 1052 potential sequence options! Each of these options would vary in the overall protein shape, as the nature of the amino acid side chains helps to determine the interaction of the protein with the other residues in the protein itself and with its surrounding environment.
The character of the amino acids throughout the protein help the protein to fold and form its 3-dimentional structure. It is this 3-D shape that is required for the functional activity of the protein (ie. protein shape = protein function). For proteins found inside the watery environments of the cell, hydrophobic amino acids will often be found on the inside of the protein structure, whereas water-loving hydrophilic amino acids will be on the surface where they can hydrogen bond and interact with the water molecules. Proline is unique because it has the only R-group that forms a cyclic structure with the amine functional group in the main chain. This cyclization is what causes proline to adopt the cis conformation rather than the trans conformation within the backbone. This shift is structure will often mean that prolines are positions where bends or directional changes occur within the protein. Methionine is unique, in that it serves as the starting amino acid for almost all of the many thousands of proteins known in nature. Cysteines contain thiol functional groups and thus, can be oxidized with other cysteine residues to form covalent disulfide bonds within the protein structure (Figure 2.14). Disulfide bridges add additional stability to the 3-D structure and are often required for correct protein folding and function (Figure 2.14).
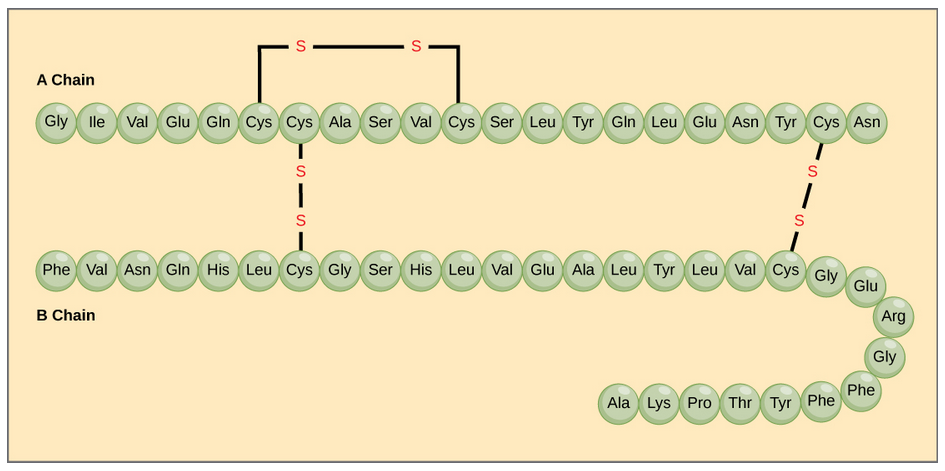
Figure 2.14 Disulfide Bonds. Disulfide bonds are formed between two cysteine residues within a peptide or protein sequence or between different peptide or protein chains. In the example above the two peptide chains that form the hormone insulin are depicted. Disulfide bridges between the two chains are required for the proper function of this hormone to regulate blood glucose levels.
Image by: CNX OpenStax via Wikimedia Commons
Protein Shape and Function
The primary structure of each protein leads to the unique folding pattern that is characteristic for that specific protein. In summary, the primary sequence is the linear order of the amino acids as they are linked together in the protein chain (Figure 2.15). In the next section, we will discuss protein folding that gives rise to secondary, tertiary and sometimes quaternary protein structures.
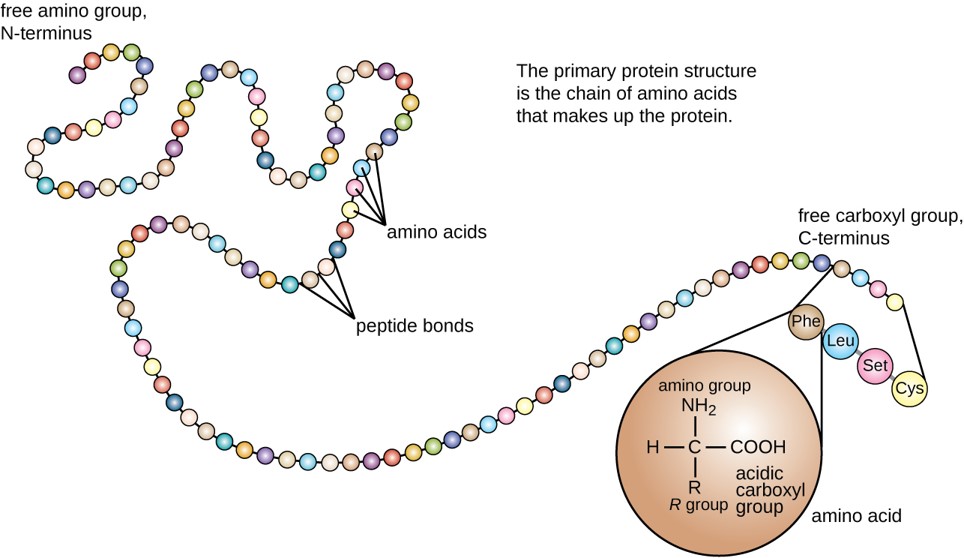
Figure 2.15 Primary protein structure is the linear sequence of amino acids.
(credit: modification of work by National Human Genome Research Institute)
back to the top
2.3 Secondary Protein Structure
In the previous section, we noted the rigidity created by the C-N bond in the amide linkage when amino acids are joined with one another and learned that this causes the amino acid R-groups to favor the trans confromation (except for proline which favors the cis conformation). This rigidity with the protein backbone limits the folding potential and patterns of the resulting protein. However, the bonds attached to the α-carbon can freely rotate and contribute to the flexibility and unique folding patterns seen within proteins. To evaluate the possible rotation patterns that can arise around the α-carbon, the torsion angles Phi (Φ) and Psi (ψ) are commonly measured. The torsion angle Phi (Φ) measures the rotation around the α-carbon – nitrogen bond by evaluating the angle between the two neighboring carbonyl carbons when you are looking directly down the α-carbon – nitrogen bond into the plane of the paper (Figure 2.16). Conversely, the torsion angle Psi (ψ) measures the rotation around the α-carbon – carbonyl carbon bond by evaluating the angle between the two neighboring nitrogen atoms when you are looking directly down the α-carbon – carbonyl carbon bond (Figure 2.16).
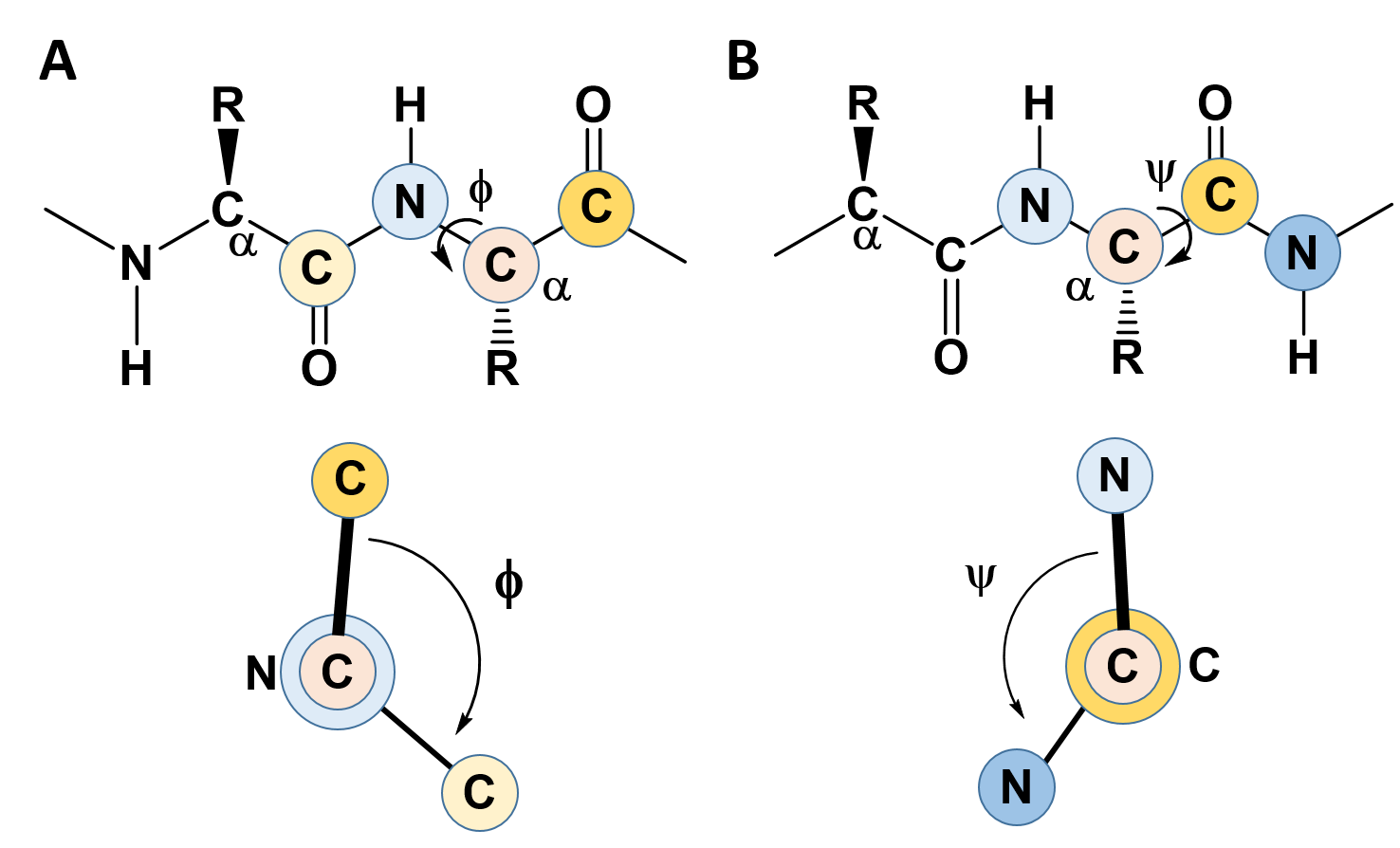
Figure 2.16 Phi (Φ) and Psi (ψ) Torsion Angles. (A) The Phi (Φ) torsion angle is a measure of the rotation around the bond between the α-carbon and the amide nitrogen. It is measured as the angle between the two carbonyl carbon atoms adjacent to the bond, shown in the lower panel. (B) The Psi (ψ) torsion angle is a measure of the rotation around the bond between the α-carbon and the carbonyl carbon. It is measured as the angle between the two nitrogen atoms adjacent to the bond, shown in the lower panel.
While the bonds around the α-carbon can rotate freely, the favored torsion angles are limited to a smaller subset of possibilities as neighboring atoms avoid conformations that have high steric hindrance associated with them. G.N. Ramachandran created computer models of small peptides to determine the stable conformations of the Phi (Φ) and Psi (ψ) torsion angles. With his results, he created what is known as the Ramachandran Plot, which graphically displays the overlap regions of the most favorable Phi (Φ) and Psi (ψ) torsion angles (Figure 2.17)
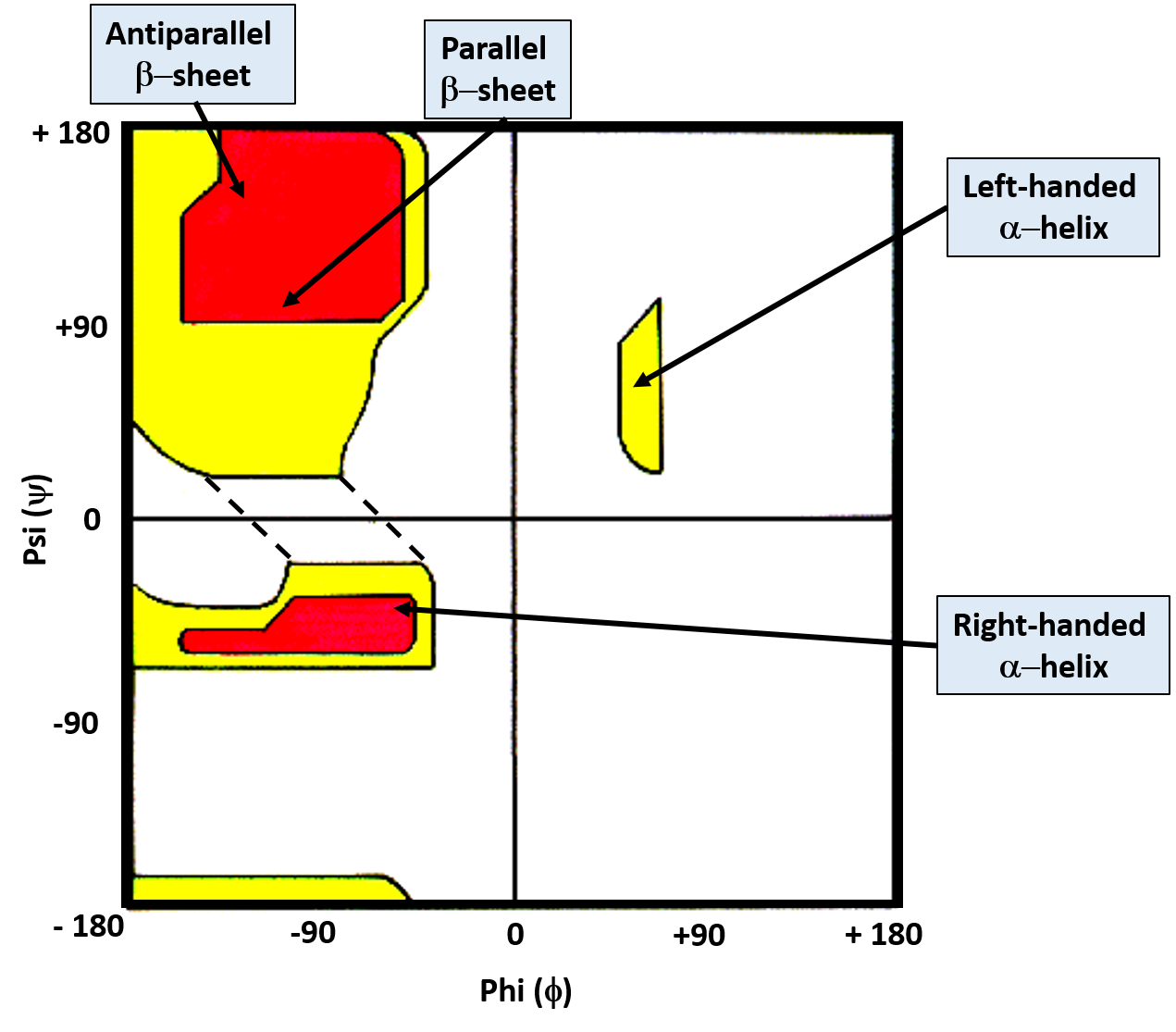
Figure 2.17 The Ramachandran Plot. Favorable and highly favorable Phi (Φ) and Psi (ψ) torsion angles are indicated in yellow and red, respectively. Bond angles for common secondary protein structures are indicated.
Image modified from: J. Cooper
Within each protein small regions of the protein may adopt specific, repeating folding patterns. These specific motifs or patterns are called secondary structure. Two of the most common secondary structural features include alpha helix and beta-pleated sheet (Figure 2.18). Within these structures, intramolecular interactions, especially hydrogen bonding between the backbone amine and carbonyl functional groups are critical to maintain 3-dimensional shape.
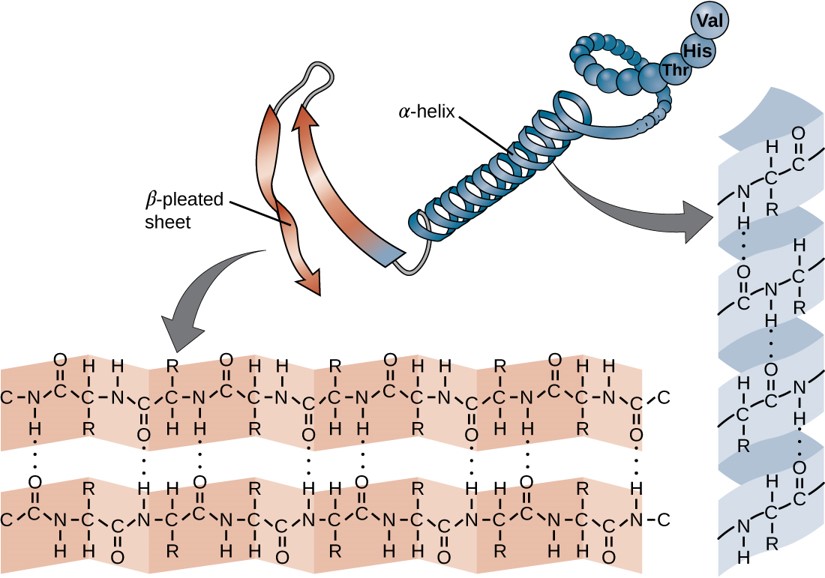
Figure 2.18 Secondary Structural Features in Protein Structure. The right-handed alpha helix and beta-pleated sheet are common structural motifs found in most proteins. They are held together by hydrogen bonding between the amine and the carbonyl oxygen within the amino acid backbone.
Image modified from: The School of Biomedical Sciences Wiki
The Alpha Helix
For the alpha helical structures, the right-handed helix is very common, whereas left-handed helices are very rare. This is due to the Phi (Φ) and Psi (ψ) torsion angles required to obtain the left-handed alpha helical structure. The protein would have to fold and twist through many unfavorable angles before obtaining the correct orientation for the left-handed helix. Thus, they are not very common in nature.
For the right-handed alpha helix, every helical turn has 3.6 amino acid residues (Figure 2.19). The R groups (the variant groups) of the polypeptide protrude out from the α-helix chain. The polypeptide backbone forms a repeating helical structure that is stabilized by hydrogen bonds between a carbonyl oxygen and an amine hydrogen. These hydrogen bonds occur at regular intervals of one hydrogen bond every fourth amino acid and cause the polypeptide backbone to form a helix. Each amino acid advances the helix, along its axis, by 1.5 Å. Each turn of the helix is composed of 3.6 amino acids; therefore the pitch of the helix is 5.4 Å. There is an average of ten amino acid residues per helix. Different amino acids have different propensities for forming α-helix. Amino acids that prefer to adopt helical conformations in proteins include methionine, alanine, leucine, glutamate and lysine. Proline and glycine have almost no tendency to form helices.
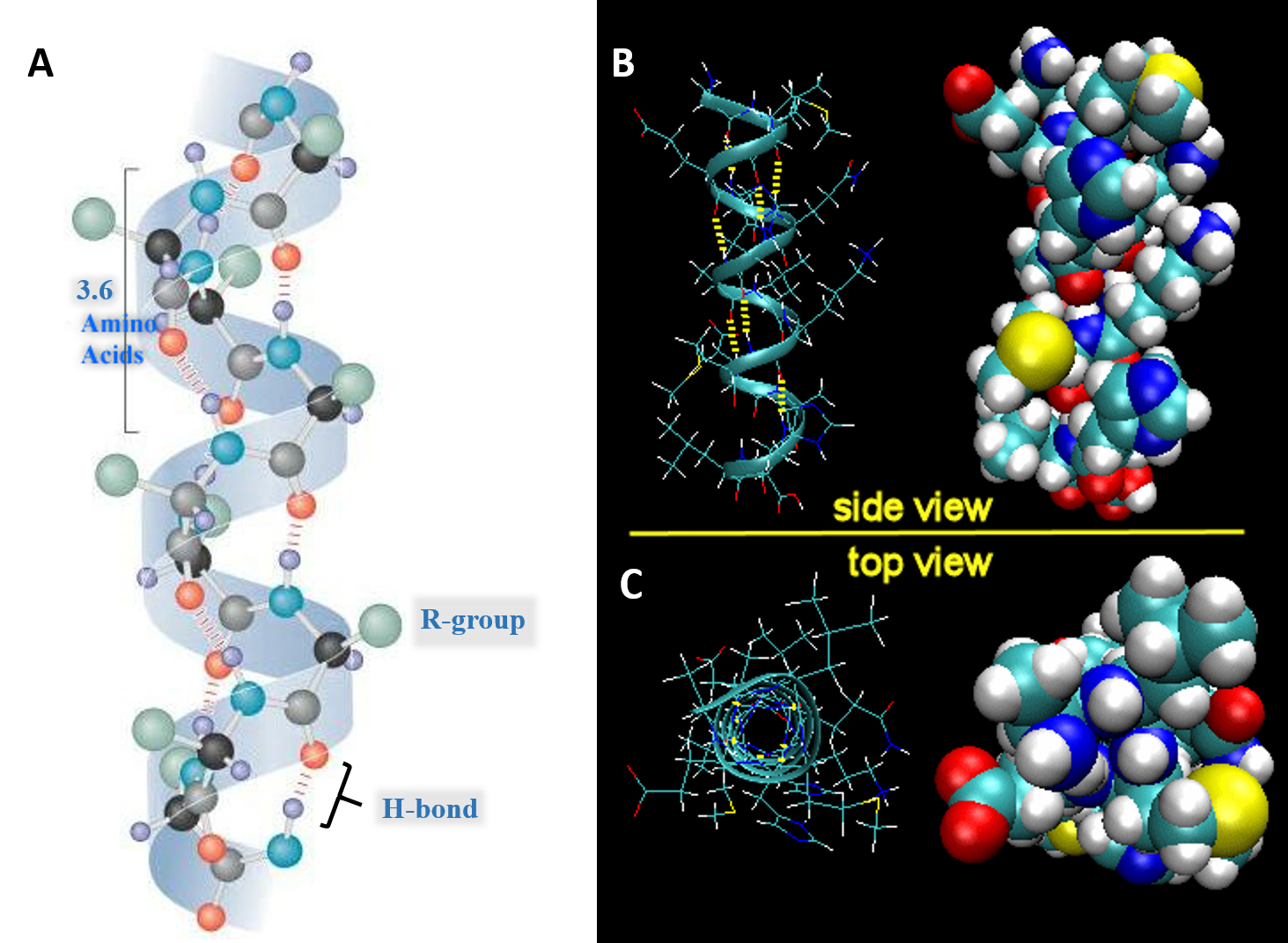
Figure 2.19 Structure of the Right-handed Alpha Helix. (A) Ball and Stick Model Side View. A total of 3.6 amino acids are required to form one turn of an α-helix. Hydrogen bonding between the carbonyl oxygen and the nitrogen of the 4th amino acid stabilize the helical structure. On the structure shown, the black atoms are the alpha carbon, grey are carbonyl carbons, red are oxygen, blue are nitrogen, green are R-groups, and light purple are hydrogen atoms. (B) Expanded Side View Linear Structure and Space-Filling Model (C) Expanded Top View Linear Structure and Space-Filling Model
Image A modified from: Maksim Image B and C from: Henry Jakubowski
Key Points about the Alpha Helix:
- The alpha helix is more compact than the fully extended polypeptide chain with phi/psi angles of 180o
- In proteins, the average number of amino acids in a helix is 11, which gives 3 turns.
- The left-handed alpha helix, although allowed from inspections of a Ramachandran plot, is rarely observed, since the amino acids used to build protein structure are L-amino acids and are biased towards forming the right-handed helix. When left-handed helices do form, they are often critical for the correct protein folding, protein stability, or are directly involved in the formation of the active site.
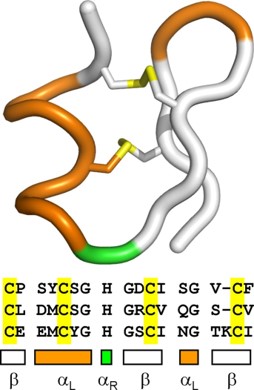
Figure 2.20 Left Handed Alpha Helix Structure. In this diagram the left handed alpha helix, shown in yellow, is part of a hairpin turn within the protein structure and is stabilized by two disulfide bridges shown in yellow.
Figure from: Annavarapu, S., Nanda, V. (2009) BMC Struct Biol 9, 61
- The core of the helix is packed tightly. There are not holes or pores in the helix.
- All the R-groups extend outward and away from the helix axis. The R-groups can be hydrophilic or hydrophobic, and can be localized in specific positions on the helix forming amphipathic regions on the protein or fully hydrophobic helices may also extend through the plasma membrane as shown in Figure 2.21
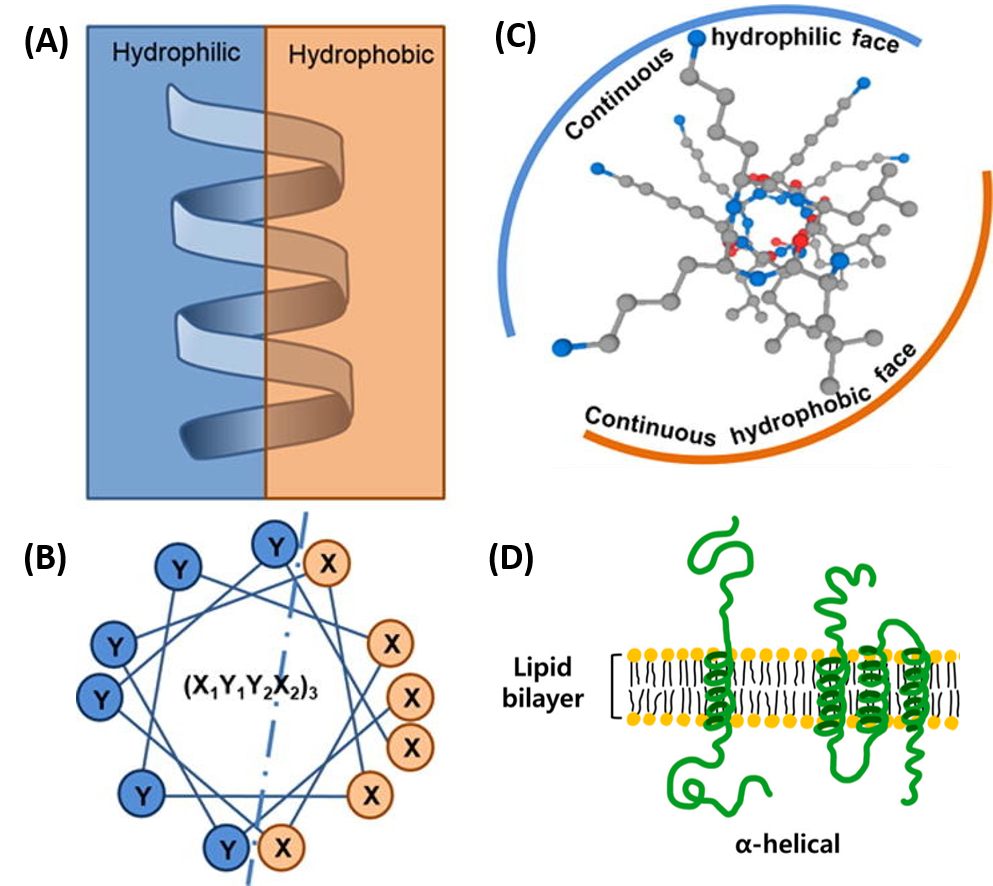
Figure 2.21 Positioning of the R-Groups within Alpha Helical Structures. R-groups may be positioned within the alpha helix to create amphipathic regions within the protein, where hydrophilic residues are positioned on one-side of the helix and hydrophobic on the other as shown in the side view (A) or top down views (B & C). R-groups may also be fully hydrophobic within alpha helices that span the plasma membrane as shown in (D).
Figure modified from: Khara, J.S., et al. (2017) Acta Biomat 57:103-114 and Ryu, H., et al. (2019) Int J Mol Sci 20 (6) 1437
- Some amino acids are more commonly found in alpha helices than other. Here are the amino acids that are typically NOT found in alpha helical structures: Gly is too small and conformationally flexible to be found with high frequency in alpha helices, while Pro is too rigid and in the cis-conformation. Pro often disrupts helical structure by causing bends in the protein. Some amino acids with side chains that can H-bond (Ser, Asp, and Asn) and aren’t too long appear to act as competitors of main chain H bond donor and acceptors, and destabilize alpha helices. Early branching R-groups, such as Val and Ile, destabilize the alpha helix due to steric interactions of the bulky side chains with the helix backbone.
- Summary of amino acids propensities for alpha helices (and beta structure as well)
- Alpha keratins, the major component of hair, skin, fur, beaks, and fingernails, are almost all alpha helix.
![]() Jmol: Updated An isolated helix from an Antifreeze Protein Jmol14 (Java) | JSMol (HTML5)
Jmol: Updated An isolated helix from an Antifreeze Protein Jmol14 (Java) | JSMol (HTML5)
The Beta Pleated Sheet:
In the β-pleated sheet, the “pleats” are formed by hydrogen bonding between atoms on the backbone of the polypeptide chain. The R groups are attached to the carbons and extend above and below the folds of the pleat in the trans conformation. The pleated segments align parallel or antiparallel to each other, and hydrogen bonds form between the partially positive nitrogen atom in the amino group and the partially negative oxygen atom in the carbonyl group of the peptide backbone (Figure 2.21).
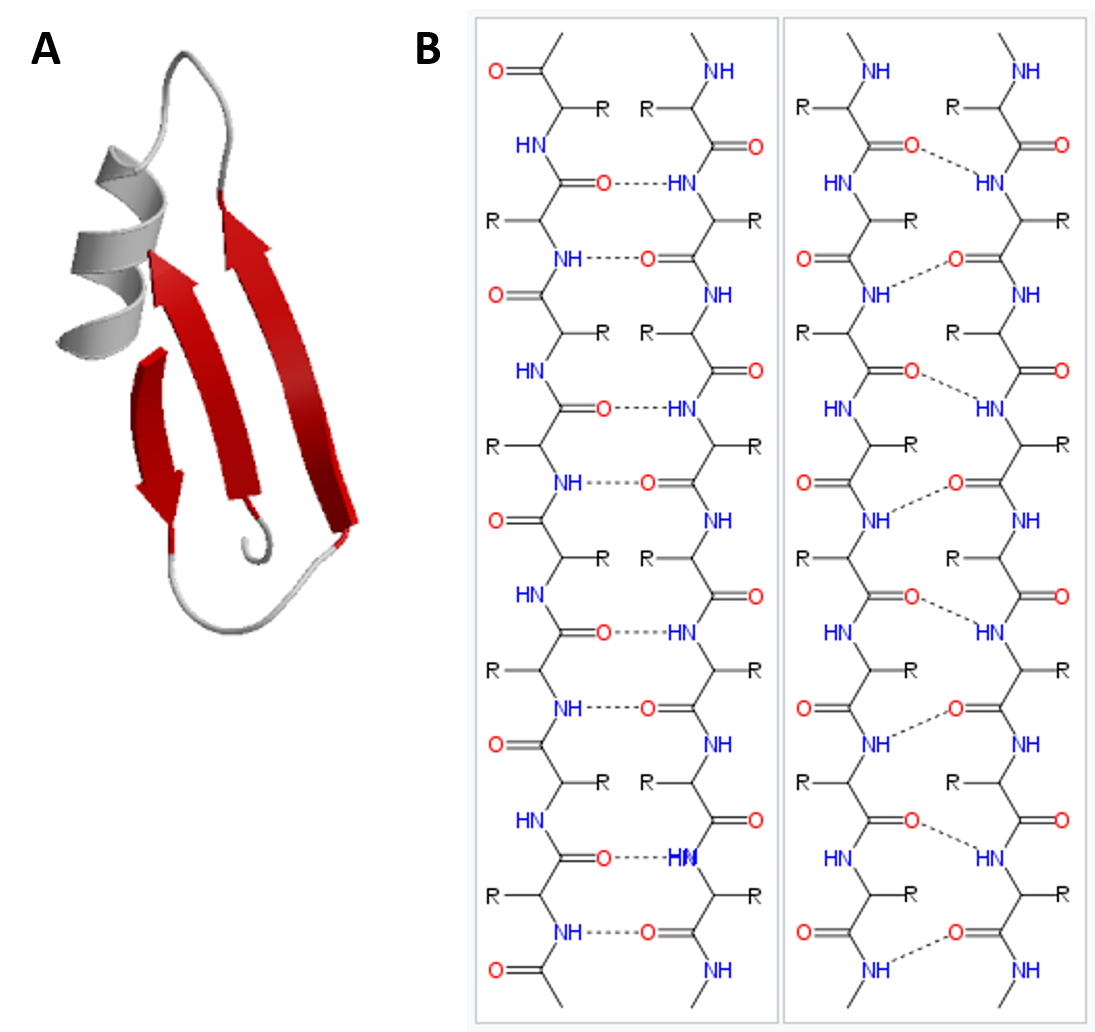
Figure 2.21 Beta-Pleated Sheet Structure. The β-pleated sheet can be oriented in the parallel or antiparallel orientation, shown in (A) above with the β-pleated sheet represented by the red ribbon arrows. The direction of the arrow indicated the orientation of the protein with the arrow running in the N- to C- direction. Hydrogen bonding between the backbone carbonyl and the backbone amine functional groups stabilized both the antiparallel (B left) and the parallel (B right) β-pleated sheet structures.
Image (A) from: Xenoblast Image (B) from: Fvasconcellos
Other Secondary Structure Motifs:
Other important secondary structures include turns, loops, hairpins and flexible linkers. There are many different classifications of turns within protein structure, including α-turns, β-turns, γ-turns, δ-turns and π-turns. β-turns (the most common form) typically contain four amino acid residues (Fig 2.22). Proline and Glycine are commonly found in turn motifs, as the cis conformation of Proline favors sharper conformational bends, while the minimal Glycine side chain allows for tighter packing of the amino acids to favor the turn structure.
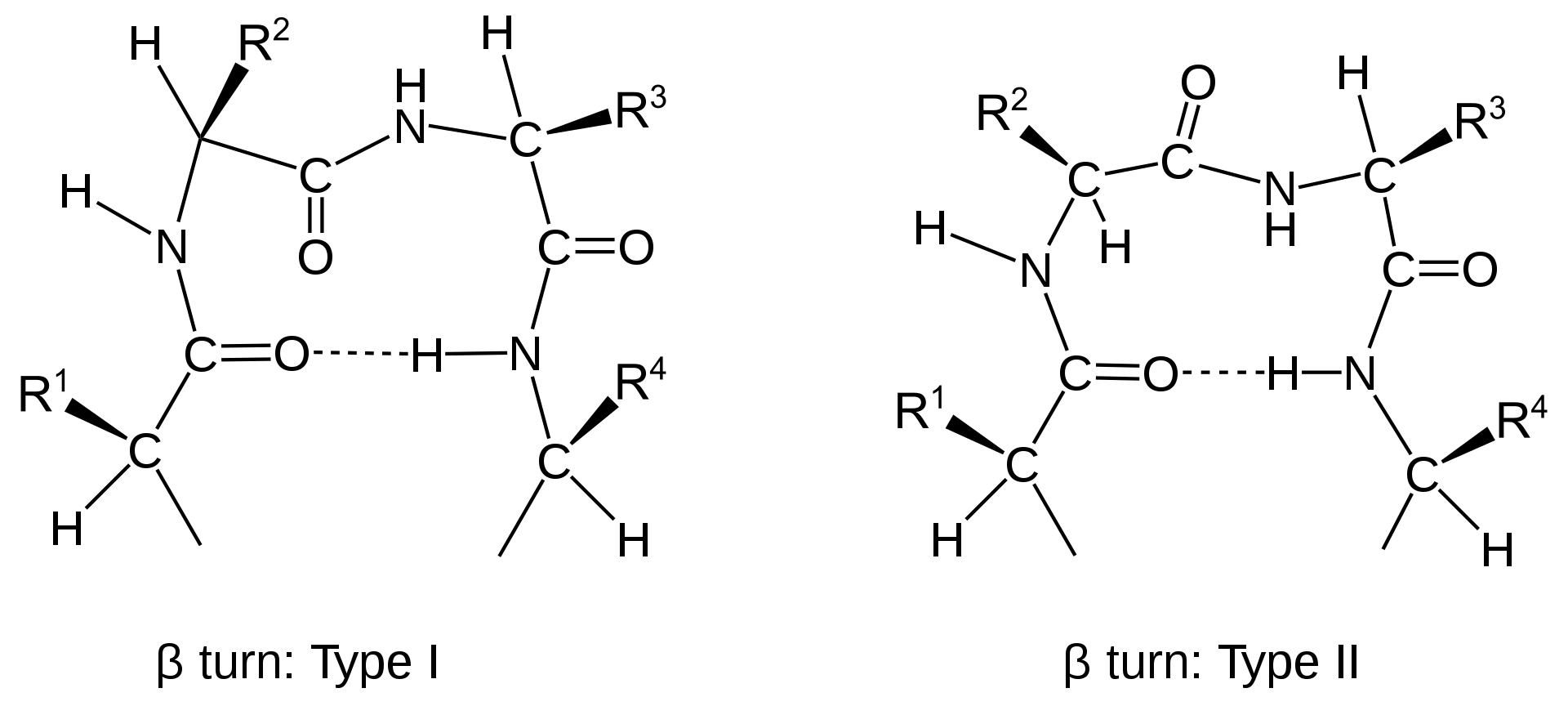
Figure 2.22 Schematic of Type I and II β-turns.
Image from: Muskid
An ω-loop is a catch-all term for a longer, extended or irregular loop without fixed internal hydrogen bonding. A hairpin is a special case of a turn, in which the direction of the protein backbone reverses and the flanking secondary structure elements interact. For example, a beta hairpin connects two hydrogen-bonded, antiparallel β-strands. Turns are sometimes found within flexible linkers or loops connecting protein domains. Linker sequences vary in length and are typically rich in polar uncharged amino acids. Flexible linkers allow connecting domains to freely twist and rotate to recruit their binding partners via protein domain dynamics.
back to the top
2.4 Supersecondary Structure and Protein Motifs
In between the secondary structure and tertiary structure of proteins are larger 3-dimensional features that have been identified in multiple different protein structures. They are known as supersecondary structure and as protein motifs. Supersecondary structure is usually composed of two secondary structures linked together by a turn and includes helix-turn-helix, helix-loop-helix, α-α corners, β-β corners, and β-hairpin-β (Figure 2.23).
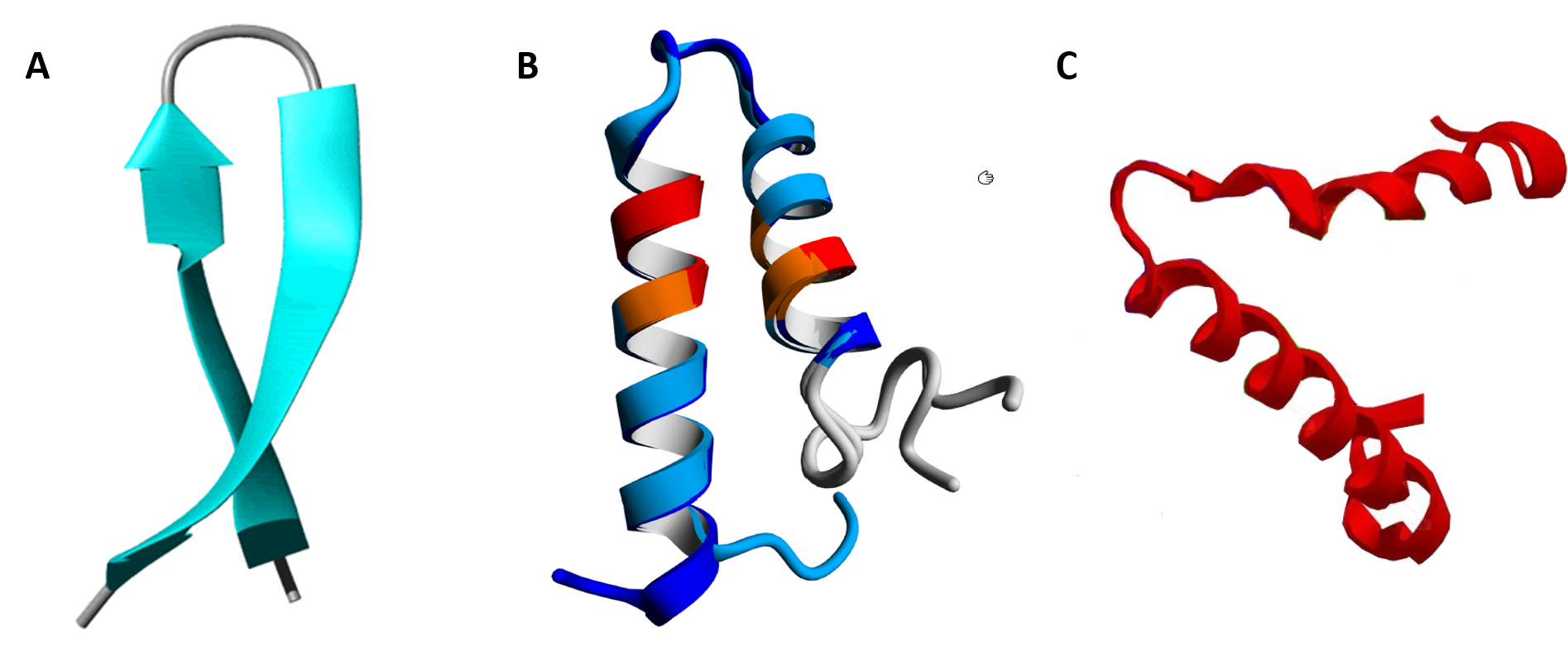
Figure 2.23 Examples of Supersecondary Structures. (A) β-hairpin-β structures are characterized by a sharp hairpin turn that does not disrupt the hydrogen bonding of the two β-pleated sheet structures. (B) Proposed helix-turn-helix structure of the Taspase1 protein, (C) α-α corner structure present in the Myoglobin protein.
Image A by: Isabella Daidone Image B by: Johannes van den Boom, et al. (2016) PLosONE 11(3):e0151431
Image C modified from: Belles14104
Protein motifs are more complex structures created from secondary and supersecondary structural components that are repeated modalities visualized in many protein structures.
Beta strands have a tendency to twist in the right hand direction to help minimize conformational energy. This leads to the formation of interesting structural motifs found in many types of proteins. Two of these structures include twisted sheets or saddles as well as beta barrels (Figure 2.24)
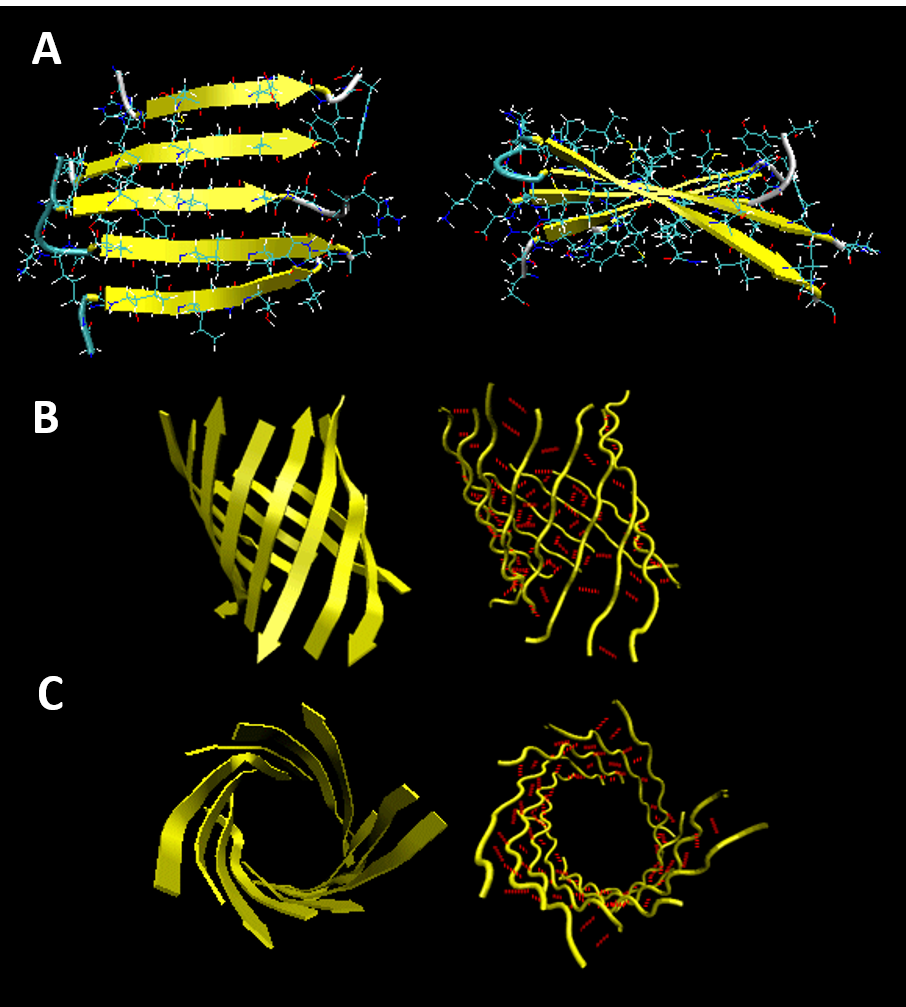
Figure 2.24 Common Beta Strand Structural Motifs. (A) Right-handed Twisted Sheet Top and Side View, (B) Beta Barrel Side View, and (C) Beta Barrel Top View
Image by: Henry Jakubowski
Structural motifs can serve particular functions within proteins such as enabling the binding of substrates or cofactors. For example, the Rossmann fold is responsible for binding to nucleotide cofactors such as nicotinamide adenine dinucleotide (NAD+) (Figure 2.25). The Rossmann fold is composed of six parallel beta strands that form an extended beta sheet. The first three strands are connected by α-helices resulting in a beta-alpha-beta-alpha-beta structure. This pattern is duplicated once to produce an inverted tandem repeat containing six strands. Overall, the strands are arranged in the order of 321456 (1 = N-terminal, 6 = C-terminal). Five stranded Rossmann-like folds are arranged in the order 32145. The overall tertiary structure of the fold resembles a three-layered sandwich wherein the filling is composed of an extended beta sheet and the two slices of bread are formed by the connecting parallel alpha helices.
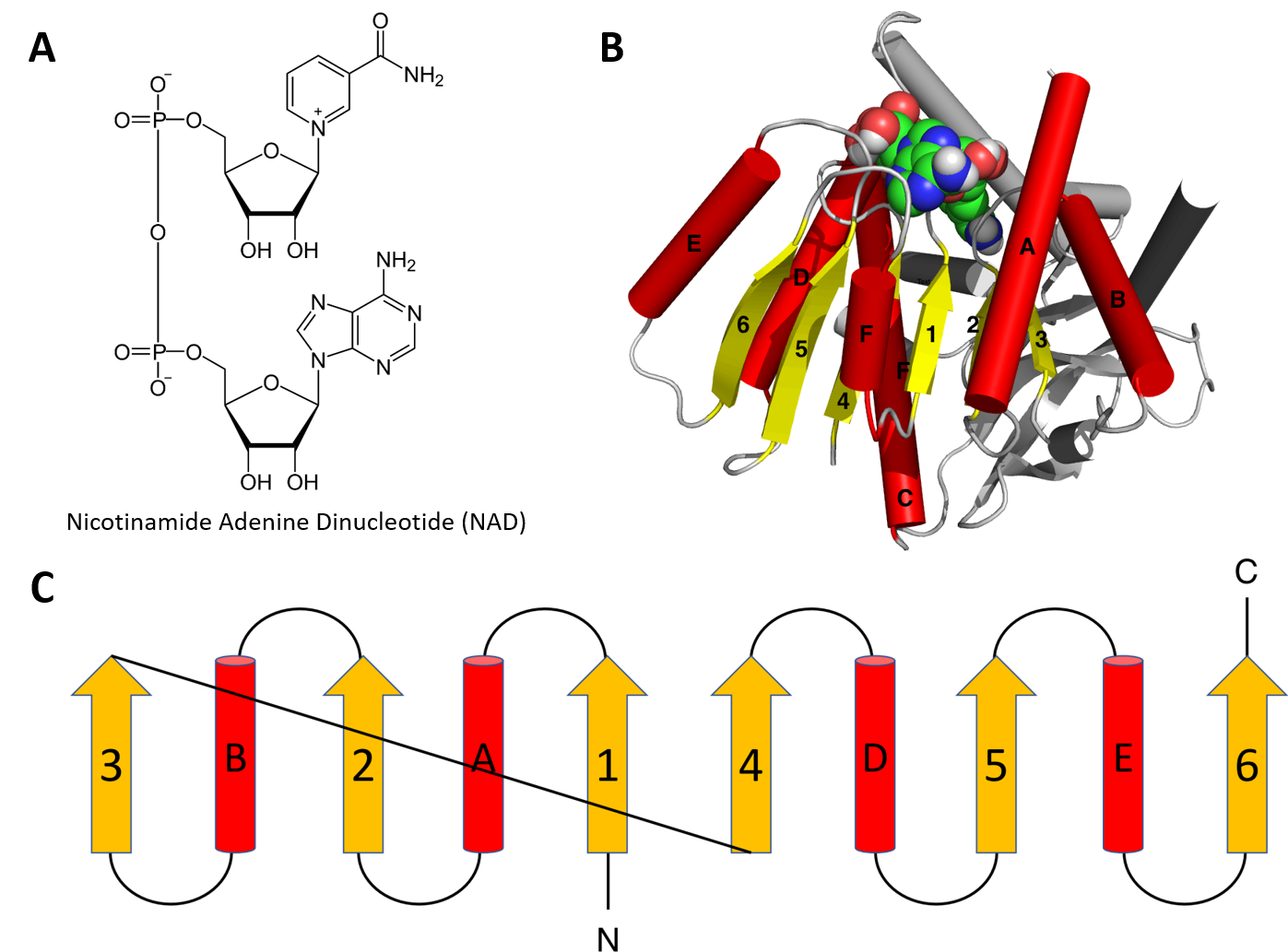
Figure 2.25 The Rossman Fold. (A) Structure of Nicotinamide Adenine Dinucleotide (NAD+) (B) Cartoon diagram of the Rossmann Fold (helices A-F red and strands 1-6 yellow) from E. coli malate dehydrogenase enzyme. The NAD+ cofactor is shown binding as the space filling molecule. (C) Schematic diagram of the six stranded Rossmann fold.
Image modified from: Boghog
One of the features if the Rossmann fold is its co-factor binding specificity. The most conserved segment of Rossmann folds is the first beta-alpha-beta segment. Since this segment is in contact with the ADP portion of dinucleotides such as FAD, NAD and NADP it is also called as an “ADP-binding beta-beta fold”.
Interestingly, similar structural motifs do not always have a common evolutionary ancestor and can arise by convergent evolution. This is the case with the TIM Barrel, a conserved protein fold consisting of eight α-helices and eight parallel β-strands that alternate along the peptide backbone. The structure is named after triosephosphate isomerase, a conserved metabolic enzyme. TIM barrels are one of the most common protein folds. One of the most intriguing features among members of this class of proteins is although they all exhibit the same tertiary fold there is very little sequence similarity between them. At least 15 distinct enzyme families use this framework to generate the appropriate active site geometry, always at the C-terminal end of the eight parallel beta-strands of the barrel.
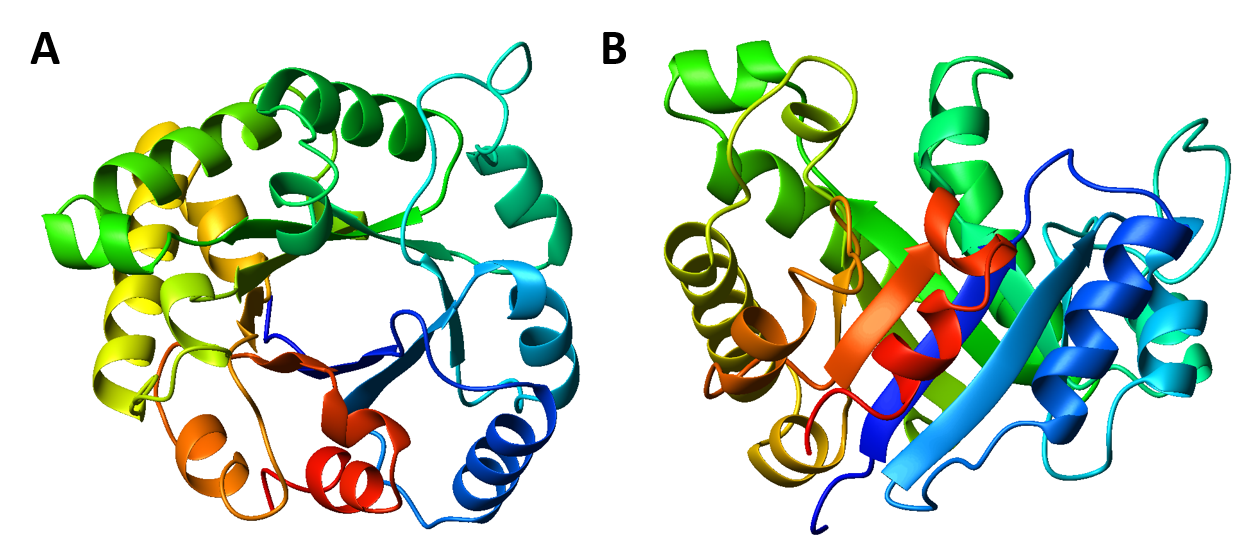
Figure 2.26 The TIM Barrel. TIM barrels are considered α/β protein folds because they include an alternating pattern of α-helices and β-strands in a single domain. In a TIM barrel the helices and strands (usually 8 of each) form a solenoid that curves around to close on itself in a doughnut shape, topologically known as a toroid. The parallel β-strands form the inner wall of the doughnut (hence, a β-barrel), whereas the α-helices form the outer wall of the doughnut. Each β-strand connects to the next adjacent strand in the barrel through a long right-handed loop that includes one of the helices, so that the ribbon N-to-C coloring in the top view (A) proceeds in rainbow order around the barrel. The TIM barrel can also be thought of, then, as made up of 8 overlapping, right-handed β-α-β super-secondary structures, as shown in the side view (B).
Image modified from: WillowW
Although the ribbon diagram of the TIM Barrel shows a hole in the protein’s central core, the amino acid side chains are not shown in this representation (Figure 2.26). The protein’s core is actually tightly packed, mostly with bulky hydrophobic amino acid residues although a few glycines are needed to allow wiggle room for the highly constrained center of the 8 approximate repeats to fit together. The packing interactions between the strands and helices are also dominated by hydrophobicity and the branched aliphatic residues valine, leucine, and isoleucine comprise about 40% of the total residues in the β-strands.
As our knowledge continues to increase about the myriad of structural motifs found in nature’s treasure trove of protein structures, we continue to gain insight into how protein structure is related to function and are better enabled to characterize newly acquired protein sequences using in silico technologies.
back to the top
2.5 Tertiary and Quaternary Protein Structure
The complete 3-dimensional shape of the entire protein (or sum of all the secondary structural motifs) is known as the tertiary structure of the protein and is a unique and defining feature for that protein (Figure 2.27). Primarily, the interactions among R groups creates the complex three-dimensional tertiary structure of a protein. The nature of the R groups found in the amino acids involved can counteract the formation of the hydrogen bonds described for standard secondary structures such as the alpha helix. For example, R groups with like charges are repelled by each other and those with unlike charges are attracted to each other (ionic bonds). Uncharged nonpolar side chains can form hydrophobic interactions. Interaction between cysteine side chains can lead to the formation of disulfide linkages.

Figure 2.27 Tertiary Protein Structure. The tertiary structure of proteins is determined by a variety of chemical interactions. These include hydrophobic interactions, ionic bonding, hydrogen bonding and disulfide linkages.
Image by: School of Biomedical Sciences Wiki
All of these interactions, weak and strong, determine the final three-dimensional shape of the protein. When a protein loses its three-dimensional shape, it is usually no longer be functional.
In nature, some proteins are formed from several polypeptides, also known as subunits, and the interaction of these subunits forms the quaternary structure. Weak interactions between the subunits help to stabilize the overall structure. For example, insulin (a globular protein) has a combination of hydrogen bonds and disulfide bonds that cause it to be mostly clumped into a ball shape. Insulin starts out as a single polypeptide and loses some internal sequences during cellular processing that form two chains held together by disulfide linkages as shown in figure 2.14. Three of these structures are then grouped further forming an inactive hexamer (Figure 2.28). The hexamer form of insulin is a way for the body to store insulin in a stable and inactive conformation so that it is available for release and reactivation in the monomer form.
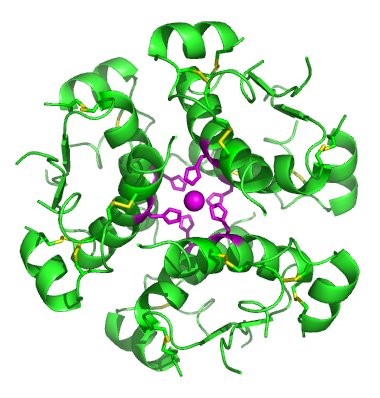
Figure 2.28 The Insulin Hormone is a Good Example of Quaternary Structure. Insulin is produced and stored in the body as a hexamer (a unit of six insulin molecules), while the active form is the monomer. The hexamer is an inactive form with long-term stability, which serves as a way to keep the highly reactive insulin protected, yet readily available.
Figure By: Isaac Yonemoto
Predicting the folding pattern of a protein based on its primary sequence is an extremely difficult task due to the inherent flexibility of amino acid residues that can be utilized to form different secondary features. As described by Fujiwara, et al., the SCOP classification (Structural Classification of Protein) and SCOPe (the extended version) are major databases providing detailed and comprehensive descriptions of all known protein structures. SCOP classification is based on hierarchical levels: The first two levels, family and superfamily, describe near and far evolutionary relationships, whereas the third, fold, describes geometrical relationships and structural motifs within the protein. Within the fold classification scheme, most proteins are assigned to one of four structural classes: (1) all α-helix, (2) all β-sheet, (3) α/β for proteins with dispersed patterns, and (4) α + β for proteins with regions that are predominated by one or the other pattern type.
Based on their shape, function and location proteins can be characterized broadly as fibrous, globular, membrane, or disordered.
Fibrous Proteins
Fibrous Proteins are characterized by elongated protein structures. These types of proteins often aggregate into filaments or bundles forming structural scaffolds in biological systems. Within animals, the two most abundant fibrous protein families are α-keratin and collagen.
α-keratin
α-keratin is the key structural element making up hair, nails, horns, claws, hooves, and the outer layer of skin. Due to its tightly wound structure, it can function as one of the strongest biological materials and has various uses in mammals, from predatory claws to hair for warmth. α-keratin is synthesized through protein biosynthesis, utilizing transcription and translation, but as the cell matures and is full of α-keratin, it dies, creating a strong non-vascular unit of keratinized tissue.
The first sequences of α-keratins were determined by Hanukoglu and Fuchs. These sequences revealed that there are two distinct but homologous keratin families which were named as Type I keratin and Type II keratins. There are 54 keratin genes in humans, 28 of which code for type I, and 26 for type II. Type I proteins are acidic, meaning they contain more acidic amino acids, such as aspartic acid, while type II proteins are basic, meaning they contain more basic amino acids, such as lysine. This differentiation is especially important in α-keratins because in the synthesis of its sub-unit dimer, the coiled coil, one protein coil must be type I, while the other must be type II (Figure 2.29). Even within type I and II, there are acidic and basic keratins that are particularly complementary within each organism. For example, in human skin, K5, a type II α-keratin, pairs primarily with K14, a type I α-keratin, to form the α-keratin complex of the epidermis layer of cells in the skin.
Coiled-coil dimers then assemble into protofilaments, a very stable, left-handed superhelical motif which further multimerises, forming filaments consisting of multiple copies of the keratin monomers (Figure 2.29). The major force that keeps the coiled-coil structures associated with one another are hydrophobic interactions between apolar residues along the keratins helical segments.
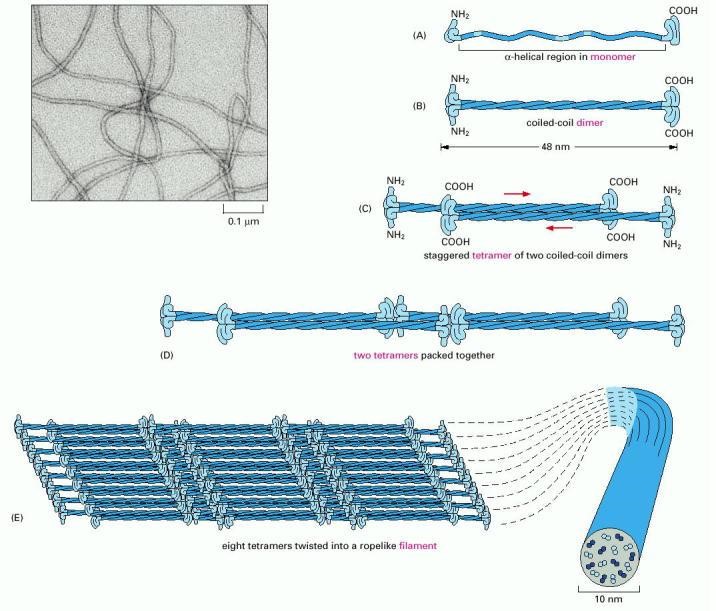
Figure 2.29. Formation of an Intermediate Filament. Intermediate filaments are composed of an α-keratin superhelical complex. Initially, two keratin monomers (A) form a coiled coil dimer structure (B) Two coiled coil dimers join to form a staggered tetramer (C), the tetramers start to join together (D), ultimately forming a sheet of eight tetramers (E). The sheet of eight tetramers is then twisted into a lefthanded helix forming the final intermediate filament (E) An electron micrograph of the intermediate filament is shown in the upper lefthand corner.
Image by: US Gov
Collagen
The fibrous protein, Collagen is the most abundant protein in mammals, making 25% to 35% of the whole-body protein content. It is found predominantly in the extracellular space within various connective tissues in the body. Collagen contains a unique quaternary structure of three protein strands wound together to form a triple helix. It is mostly found in fibrous tissues such as tendons, ligaments, and skin.
Depending upon the degree of mineralization, collagen tissues may be rigid (bone), compliant (tendon), or have a gradient from rigid to compliant (cartilage). It is also abundant in corneas, blood vessels, the gut, intervertebral discs, and the dentin in teeth. In muscle tissue, it serves as a major component of the endomysium. Collagen constitutes one to two percent of muscle tissue and accounts for 6% of the weight of strong, tendinous, muscles. The fibroblast is the most common cell that creates collagen. Gelatin, which is used in food and industry, is collagen that has been irreversibly hydrolyzed. In addition, partially and fully hydrolyzed collagen powders are used as dietary supplements. Collagen has many medical uses in treating complications of the bones and skin.
The name collagen comes from the Greek (kólla), meaning “glue”, and suffix -gen, denoting “producing”. This refers to the compound’s early use in the process of boiling the skin and tendons of horses and other animals to obtain glue.
Over 90% of the collagen in the human body is type I. However, as of 2011, 28 types of collagen have been identified, described, and divided into several groups according to the structure they form. The five most common types are:
- Type I: skin, tendon, vasculature, organs, bone (main component of the organic part of bone)
- Type II: cartilage (main collagenous component of cartilage)
- Type III: reticulate (main component of reticular fibers), commonly found alongside type I
- Type IV: forms basal lamina, the epithelium-secreted layer of the basement membrane
- Type V: cell surfaces, hair, and placenta
Here we will focus on the unique attributes of Collagen Type I. Collagen Type I has an unusual amino acid composition and sequence:
- Glycine is found at almost every third residue.
- Proline makes up about 17% of collagen.
- Collagen contains two uncommon derivative amino acids not directly inserted during translation. These amino acids are found at specific locations relative to glycine and are modified post-translationally by different enzymes, both of which require vitamin C as a cofactor (Figure 2.30).
- Hydroxyproline derived from proline
- Hydroxylysine derived from lysine – depending on the type of collagen, varying numbers of hydroxylysines are glycosylated (mostly having disaccharides attached).

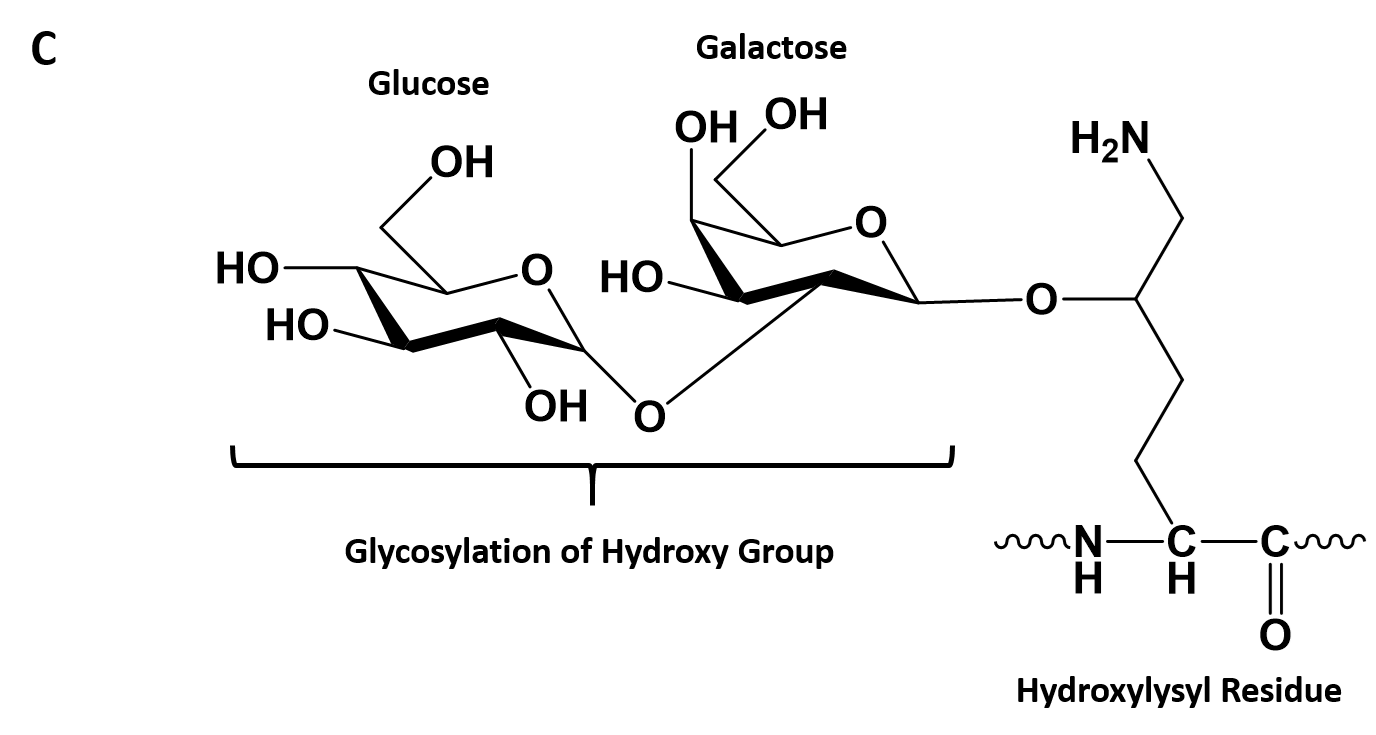
Figure 2.30. Hydroxylation of Proline and Lysine During the Post-Translational Modification of Collagen Type I. The enzymes prolyl hydroxylase and lysyl hydroxylase are required for the hydroxylation of proline (A) and lysine (B) residues, respectively. (Note: While position 3 is shown above, prolyl residues may alternatively be hydroxylated at the 4-position). The hydroxylase enzymes modify amino acid residues after they have been incorporated into the protein as a post-translational modification and require vitamin C (ascorbate) as a cofactor. (C) Further modification of the hydroxylysine residues by glycosylation can lead to the incorporation of the disaccharide (galactose-glucose) at the hydroxy oxygen.
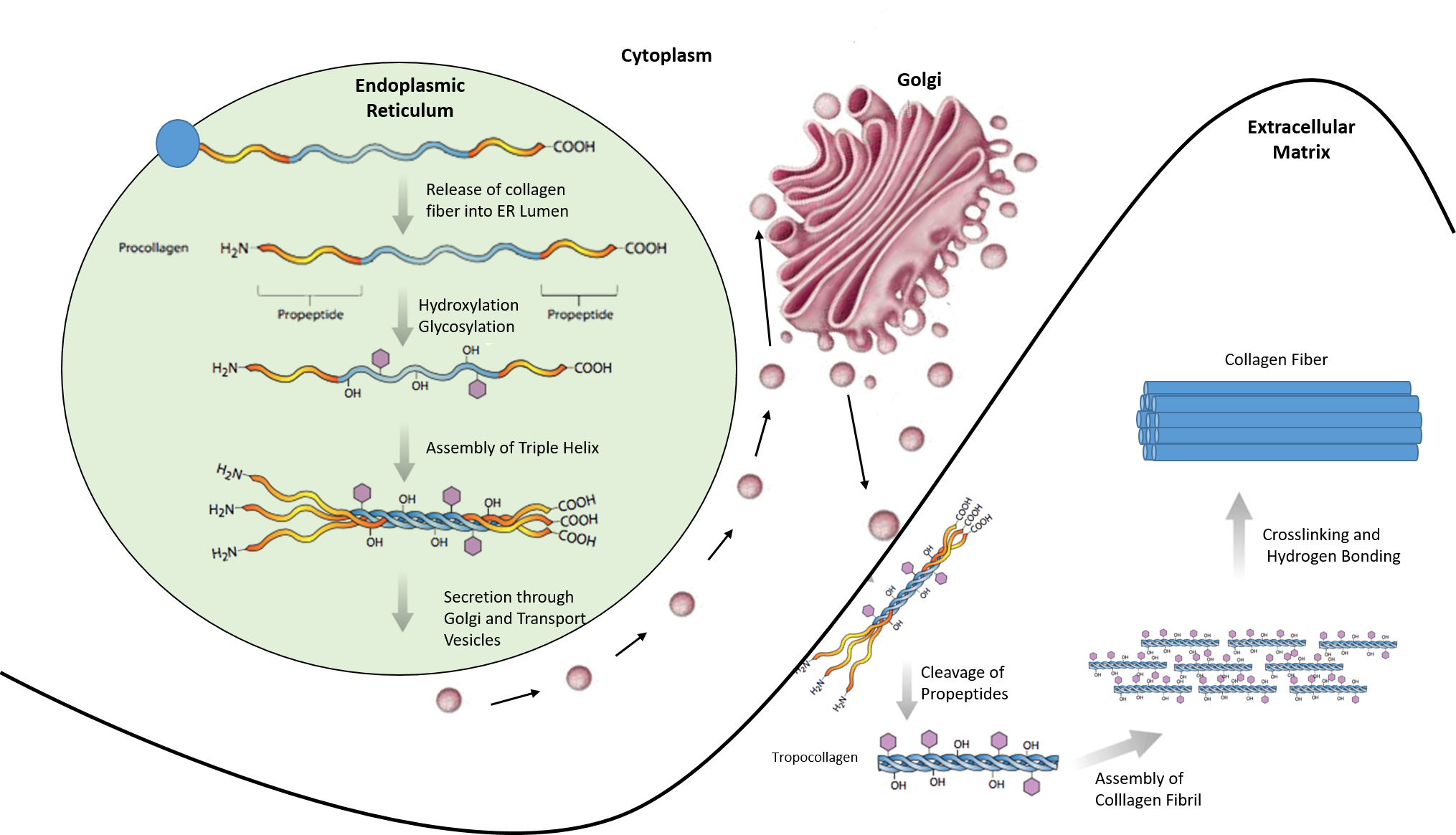
Most collagen forms in a similar manner. The synthesis process for Collagen Type I is described below and showcases the complexity of protein folding and processing (Figure 2.31).
- Inside the cell
- Two types of alpha chains are formed during translation on ribosomes along the rough endoplasmic reticulum (RER): alpha-1 and alpha-2 chains. These peptide chains (known as preprocollagen) have registration peptides on each end and a signal peptide.
- Polypeptide chains are released into the lumen of the RER.
- Signal peptides are cleaved inside the RER and the chains are now known as pro-alpha chains.
- Hydroxylation of lysine and proline amino acids occurs inside the lumen. This process is dependent on ascorbic acid (vitamin C) as a cofactor.
- Glycosylation of specific hydroxylysine residues occurs.
- Triple alpha helical structure is formed inside the endoplasmic reticulum from two alpha-1 chains and one alpha-2 chain.
- Procollagen is shipped to the Golgi apparatus, where it is packaged and secreted by exocytosis.
- Outside the cell
- Registration peptides are cleaved and tropocollagen is formed by procollagen peptidase.
- Multiple tropocollagen molecules form collagen fibrils, via covalent cross-linking (aldol reaction) by lysyl oxidase which links hydroxylysine and lysine residues. Multiple collagen fibrils form into collagen fibers.
- Collagen may be attached to cell membranes via several types of protein, including fibronectin, laminin, fibulin and integrin.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
#/media/File:Beta_turn.svg){kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 2.31. Synthesis of Collagen Type I. Polypeptide chains are synthesized in the endoplasmic reticulum and released into the lumen where they are hydroxylated and glycosylated. The procollagen triple helix is formed and transported through the golgi apparatus where it is further processed. Procollagen is secreted into the extracellular matrix where it is cleaved into tropocollagen. Tropocollagen assembles into a collagen fibril where crosslinking and hydrogen bonding occur to form the final collagen fiber.
Image modified from: E.V. Wong and Encyclopedia Britannica
{kind=link}
Vitamin C deficiency causes scurvy, a serious and painful disease in which defective collagen prevents the formation of strong connective tissue. Gums deteriorate and bleed, with loss of teeth; skin discolors, and wounds do not heal. Prior to the 18th century, this condition was notorious among long-duration military, particularly naval, expeditions during which participants were deprived of foods containing vitamin C.
An autoimmune disease such as lupus erythematosus or rheumatoid arthritis may attack healthy collagen fibers. Cortisol stimulates degradation of collagen into amino acids, suggesting that stress can worsen these disease states.
Many bacteria and viruses secrete virulence factors, such as the enzyme collagenase, which destroys collagen or interferes with its production.
back to the top
Globular Proteins
Globular proteins or spheroproteins are spherical (“globe-like”) proteins and are one of the common protein types. Globular proteins are somewhat water-soluble (forming colloids in water), unlike the fibrous or membrane proteins. There are multiple fold classes of globular proteins, since there are many different architectures that can fold into a roughly spherical shape.
The term globin can refer more specifically to proteins including the globin fold. The globin fold is a common three-dimensional fold in proteins and defines the globin-like protein superfamily (Figure 2.32). This fold typically consists of eight alpha helices, although some proteins have additional helix extensions at their termini. The globin fold is found in its namesake globin protein families: hemoglobins and myoglobins, as well as in phycocyanins. Because myoglobin was the first protein whose structure was solved, the globin fold was thus the first protein fold discovered. Since the globin fold contains only helices, it is classified as an all-alpha protein fold.

Figure 2.32 The Globin Fold. (A) An example of the globin fold, the oxygen-carrying protein myoglobin (PBD ID 1MBA) from the mollusc Aplysia limacina. (B) Structure of the tetrameric hemoglobin protein containing a total of four globin folds.
Image A by: Wikipedia Image B by: Zephyris
{kind=link}
The term globular protein is quite old (dating probably from the 19th century) and is now somewhat archaic given the hundreds of thousands of proteins and more elegant and descriptive structural motif vocabulary. The spherical structure is induced by the protein’s tertiary structure. The molecule’s apolar (hydrophobic) amino acids are bounded towards the molecule’s interior whereas polar (hydrophilic) amino acids are bound outwards, allowing dipole-dipole interactions with the solvent, which explains the molecule’s solubility.
Unlike fibrous proteins which play a predominant structural function, globular proteins can act as:
- Enzymes, by catalyzing organic reactions taking place in the organism in mild conditions and with a great specificity. Different esterases fulfill this role.
- Messengers, by transmitting messages to regulate biological processes. This function is done by hormones, i.e. insulin etc.
- Transporters of other molecules through membranes
- Stocks of amino acids.
- Regulatory roles are also performed by globular proteins rather than fibrous proteins.
- Structural proteins, e.g., actin and tubulin, which are globular and soluble as monomers, but polymerize to form long, stiff fibers
Many of the proteins that will be detailed in later chapters will fall into this class of proteins.
Membrane Proteins
Membrane proteins are proteins that are part of, or interact with, biological membranes. They include: 1) integral membrane proteins, which are part of or permanently anchored to the membrane, and 2) peripheral membrane proteins, which are attached temporarily to the membrane via integral proteins or the lipid bilayer. The integral membrane proteins are further classified as transmembrane proteins that span across the membrane, or integral monotopic proteins, which are to attached to only one side of the membrane.
Membrane proteins, like soluble globular proteins, fibrous proteins, and disordered proteins, are common. Symbolic of their importance in medicine, membrane proteins are the targets of over 50% of all modern medicinal drugs. It is estimated that 20–30% of all genes in most genomes encode for membrane proteins. Compared to other classes of proteins, determining membrane protein structures remains a challenge in large part due to the difficulty in establishing experimental conditions that can preserve the correct conformation of the protein in isolation from its native environment (Figure 2.33).
Membrane proteins perform a variety of functions vital to the survival of organisms:
- Membrane receptor proteins relay signals between the cell’s internal and external environments.
- Transport proteins move molecules and ions across the membrane. They can be categorized according to the Transporter Classification database.
- Membrane enzymes may have many activities, such as oxidoreductase, transferase or hydrolase.
- Cell adhesion molecules allow cells to identify each other and interact. For example, proteins involved in immune response.

{kind=link}
Integral membrane proteins are permanently attached to the membrane. Such proteins can be separated from the biological membranes only using detergents, nonpolar solvents, or sometimes denaturing agents. They can be classified according to their relationship with the bilayer:
- Integral polytopic proteins are transmembrane proteins that span across the membrane more than once. These proteins may have different transmembrane topology. These proteins have one of two structural architectures:
- helix bundle proteins, which are present in all types of biological membranes;
- beta barrel proteins, which are found only in outer membranes of Gram-negative bacteria, and outer membranes of mitochondria and chloroplasts.
- Bitopic proteins are transmembrane proteins that span across the membrane only once. Transmembrane helices from these proteins have significantly different amino acid distributions to transmembrane helices from polytopic proteins.
- Integral monotopic proteins are integral membrane proteins that are attached to only one side of the membrane and do not span the whole way across.

Figure 2.34 Schematic representation of the different types of interaction between monotopic membrane proteins and the cell membrane. 1. interaction by an amphipathic α-helix parallel to the membrane plane (in-plane membrane helix) 2. interaction by a hydrophobic loop 3. interaction by a covalently bound membrane lipid (lipidation) 4. electrostatic or ionic interactions with membrane lipids.
{kind=link}
Peripheral membrane proteins are temporarily attached either to the lipid bilayer or to integral proteins by a combination of hydrophobic, electrostatic, and other non-covalent interactions. Peripheral proteins dissociate following treatment with a polar reagent, such as a solution with an elevated pH or high salt concentrations.
Integral and peripheral proteins may be post-translationally modified, with added fatty acid, diacylglycerol or prenyl chains, or GPI (glycosylphosphatidylinositol), which may be anchored in the lipid bilayer.
Disordered Proteins
An intrinsically disordered protein (IDP) is a protein that lacks a fixed or ordered three-dimensional structure (Figure 2.35). IDPs cover a spectrum of states from fully unstructured to partially structured and include random coils, (pre-)molten globules, and large multi-domain proteins connected by flexible linkers. They constitute one of the main types of protein (alongside globular, fibrous and membrane proteins).
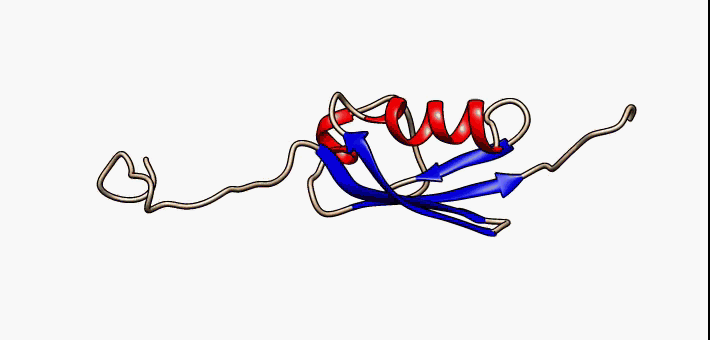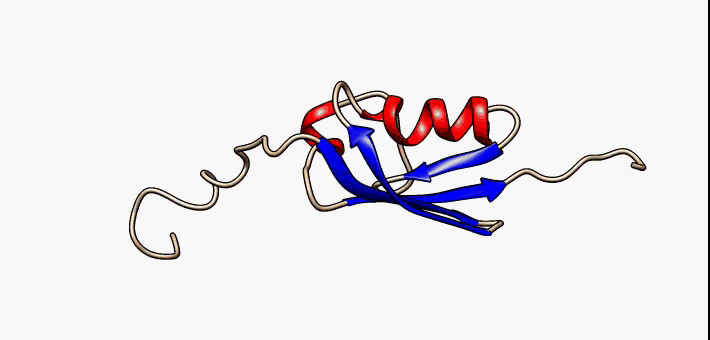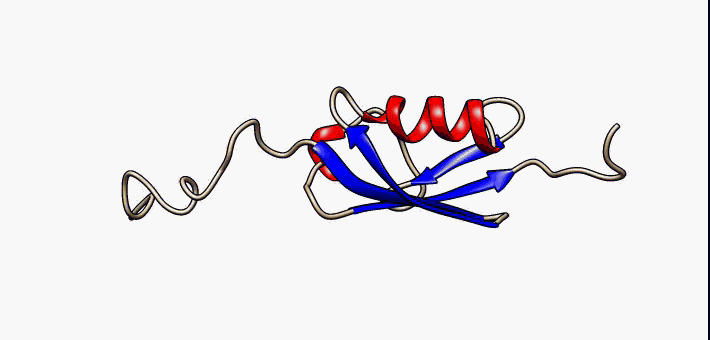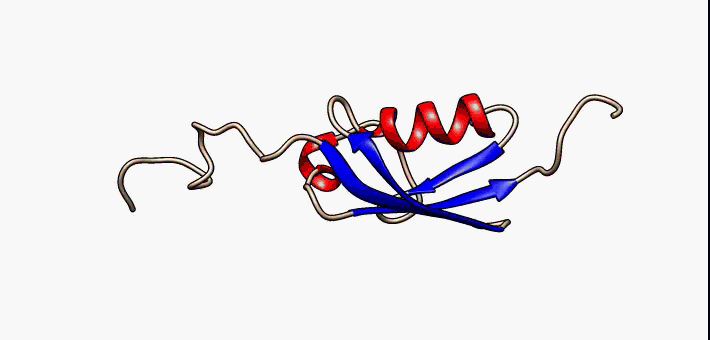
Figure 2.35 Conformational flexibility in SUMO-1 protein (PDB:1a5r). The central part shows relatively ordered structure. Conversely, the N- and C-terminal regions (left and right, respectively) show ‘intrinsic disorder’, although a short helical region persists in the N-terminal tail. Ten alternative NMR models were morphed. Secondary structure elements: α-helices (red), β-strands (blue arrows).
Image by: Lukasz Kozlowski
{kind=link}
The discovery of IDPs has challenged the traditional protein structure paradigm, that protein function depends on a fixed three-dimensional structure. This dogma has been challenged over the last twenty years by increasing evidence from various branches of structural biology, suggesting that protein dynamics may be highly relevant for such systems. Despite their lack of stable structure, IDPs are a very large and functionally important class of proteins. In some cases, IDPs can adopt a fixed three-dimensional structure after binding to other macromolecules. Overall, IDPs are different from structured proteins in many ways and tend to have distinct properties in terms of function, structure, sequence, interactions, evolution and regulation.
In the 1930s -1950s, the first protein structures were solved by protein crystallography. These early structures suggested that a fixed three-dimensional structure might be generally required to mediate biological functions of proteins. When stating that proteins have just one uniquely defined configuration, Mirsky and Pauling did not recognize that Fisher’s work would have supported their thesis with his ‘Lock and Key’ model (1894). These publications solidified the central dogma of molecular biology in that the sequence determines the structure which, in turn, determines the function of proteins. In 1950, Karush wrote about ‘Configurational Adaptability’ contradicting all the assumptions and research in the 19th century. He was convinced that proteins have more than one configuration at the same energy level and can choose one when binding to other substrates. In the 1960s, Levinthal’s paradox suggested that the systematic conformational search of a long polypeptide is unlikely to yield a single folded protein structure on biologically relevant timescales (i.e. seconds to minutes). Curiously, for many (small) proteins or protein domains, relatively rapid and efficient refolding can be observed in vitro. As stated in Anfinsen’s Dogma from 1973, the fixed 3D structure of these proteins is uniquely encoded in its primary structure (the amino acid sequence), is kinetically accessible and stable under a range of (near) physiological conditions, and can therefore be considered as the native state of such “ordered” proteins.
During the subsequent decades, however, many large protein regions could not be assigned in x-ray datasets, indicating that they occupy multiple positions, which average out in electron density maps. The lack of fixed, unique positions relative to the crystal lattice suggested that these regions were “disordered”. Nuclear magnetic resonance spectroscopy of proteins also demonstrated the presence of large flexible linkers and termini in many solved structural ensembles. It is now generally accepted that proteins exist as an ensemble of similar structures with some regions more constrained than others. Intrinsically Unstructured Proteins (IUPs) occupy the extreme end of this spectrum of flexibility, whereas IDPs also include proteins of considerable local structure tendency or flexible multidomain assemblies. These highly dynamic disordered regions of proteins have subsequently been linked to functionally important phenomena such as allosteric regulation and enzyme catalysis.
Many disordered proteins have the binding affinity with their receptors regulated by post-translational modification, thus it has been proposed that the flexibility of disordered proteins facilitates the different conformational requirements for binding the modifying enzymes as well as their receptors. Intrinsic disorder is particularly enriched in proteins implicated in cell signaling, transcription and chromatin remodeling functions.
Flexible linkers
Disordered regions are often found as flexible linkers or loops connecting domains. Linker sequences vary greatly in length but are typically rich in polar uncharged amino acids. Flexible linkers allow the connecting domains to freely twist and rotate to recruit their binding partners via protein domain dynamics. They also allow their binding partners to induce larger scale conformational changes by long-range allostery.
Linear motifs
Linear motifs are short disordered segments of proteins that mediate functional interactions with other proteins or other biomolecules (RNA, DNA, sugars etc.). Many roles of linear motifs are associated with cell regulation, for instance in control of cell shape, subcellular localisation of individual proteins and regulated protein turnover. Often, post-translational modifications such as phosphorylation tune the affinity (not rarely by several orders of magnitude) of individual linear motifs for specific interactions. Unlike globular proteins IDPs do not have spatially-disposed active pockets. Nevertheless, in 80% of IDPs (~3 dozens) subjected to detailed structural characterization by NMR there are linear motifs termed PreSMos (pre-structured motifs) that are transient secondary structural elements primed for target recognition. In several cases it has been demonstrated that these transient structures become full and stable secondary structures, e.g., helices, upon target binding. Hence, PreSMos are the putative active sites in IDPs.
Coupled folding and binding
Many unstructured proteins undergo transitions to more ordered states upon binding to their targets. The coupled folding and binding may be local, involving only a few interacting residues, or it might involve an entire protein domain. It was recently shown that the coupled folding and binding allows the burial of a large surface area that would be possible only for fully structured proteins if they were much larger. Moreover, certain disordered regions might serve as “molecular switches” in regulating certain biological function by switching to ordered conformation upon molecular recognition like small molecule-binding, DNA/RNA binding, ion interactions.
Disorder in the bound state (fuzzy complexes)
Intrinsically disordered proteins can retain their conformational freedom even when they bind specifically to other proteins. The structural disorder in bound state can be static or dynamic. In fuzzy complexes structural multiplicity is required for function and the manipulation of the bound disordered region changes activity. The conformational ensemble of the complex is modulated via post-translational modifications or protein interactions. Specificity of DNA binding proteins often depends on the length of fuzzy regions, which is varied by alternative splicing. Intrinsically disordered proteins adapt many different structures in vivo according to the cell’s conditions, creating a structural or conformational ensemble.
Therefore, their structures are strongly function-related. However, only few proteins are fully disordered in their native state. Disorder is mostly found in intrinsically disordered regions (IDRs) within an otherwise well-structured protein. The term intrinsically disordered protein (IDP) therefore includes proteins that contain IDRs as well as fully disordered proteins.
The existence and kind of protein disorder is encoded in its amino acid sequence. In general, IDPs are characterized by a low content of bulky hydrophobic amino acids and a high proportion of polar and charged amino acids, usually referred to as low hydrophobicity. This property leads to good interactions with water. Furthermore, high net charges promote disorder because of electrostatic repulsion resulting from equally charged residues. Thus disordered sequences cannot sufficiently bury a hydrophobic core to fold into stable globular proteins. In some cases, hydrophobic clusters in disordered sequences provide the clues for identifying the regions that undergo coupled folding and binding (refer to biological roles).
Many disordered proteins reveal regions without any regular secondary structure These regions can be termed as flexible, compared to structured loops. While the latter are rigid and contain only one set of Ramachandran angles, IDPs involve multiple sets of angles. The term flexibility is also used for well-structured proteins, but describes a different phenomenon in the context of disordered proteins. Flexibility in structured proteins is bound to an equilibrium state, while it is not so in IDPs. Many disordered proteins also reveal low complexity sequences, i.e. sequences with over-representation of a few residues. While low complexity sequences are a strong indication of disorder, the reverse is not necessarily true, that is, not all disordered proteins have low complexity sequences. Disordered proteins have a low content of predicted secondary structure.
back to the top
2.6 Protein Folding, Denaturation and Hydrolysis
Protein folding is the physical process by which a protein chain acquires its native 3-dimensional structure, a conformation that is usually biologically functional, in an expeditious and reproducible manner (Figure 2.36). It is the physical process by which a polypeptide folds into its characteristic and functional three-dimensional structure from random coil. Each protein exists as an unfolded polypeptide or random coil when translated from a sequence of mRNA to a linear chain of amino acids. This polypeptide lacks any stable (long-lasting) three-dimensional structure (the left hand side of the first figure). As the polypeptide chain is being synthesized by a ribosome, the linear chain begins to fold into its three-dimensional structure. Folding begins to occur even during translation of the polypeptide chain. Amino acids interact with each other to produce a well-defined three-dimensional structure, the folded protein (the right hand side of the figure), known as the native state. The resulting three-dimensional structure is determined by the amino acid sequence or primary structure (Anfinsen’s dogma).
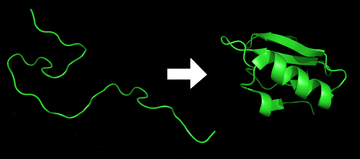
Figure 2.36 Protein Before and After Folding
Image by: DrKjaergaard
{kind=link}
The correct three-dimensional structure is essential to function, although some parts of functional proteins may remain unfolded or as in the case of IDPs remain flexible, so that protein dynamics is important. Failure to fold into native structure generally produces inactive proteins, but in some instances misfolded proteins have modified or toxic functionality. Several neurodegenerative and other diseases are believed to result from the accumulation of misfolded proteins, such as amyloid fibrils found in Alzheimer’s patients.
Folding is a spontaneous process that is mainly guided by hydrophobic interactions, formation of intramolecular hydrogen bonds, van der Waals forces, and it is opposed by conformational entropy. The process of folding often begins co-translationally, so that the N-terminus of the protein begins to fold while the C-terminal portion of the protein is still being synthesized by the ribosome; however, a protein molecule may fold spontaneously during or after biosynthesis. While these macromolecules may be regarded as “folding themselves”, the process also depends on the solvent (water or lipid bilayer), the concentration of salts, the pH, the temperature, the possible presence of cofactors and of molecular chaperones. Proteins will have limitations on their folding abilities by the restricted bending angles or conformations that are possible, as described by the Ramachandran plot.

Figure 2.37 Hydrophobic collapse. In the compact fold (to the right), the hydrophobic amino acids (shown as black spheres) collapse toward the center to become shielded from aqueous environment.
Image by: Tomixdf
{kind=link}
Protein folding must be thermodynamically favorable within a cell in order for it to be a spontaneous reaction. Since it is known that protein folding is a spontaneous reaction, then it must assume a negative Gibbs free energy value. Gibbs free energy in protein folding is directly related to enthalpy and entropy. For a negative ΔG to arise and for protein folding to become thermodynamically favorable, then either enthalpy, entropy, or both terms must be favorable.
Minimizing the number of hydrophobic side-chains exposed to water is an important driving force behind the folding process. The hydrophobic effect (Figure 2.37) is the phenomenon in which the hydrophobic chains of a protein collapse into the core of the protein (away from the hydrophilic environment). In an aqueous environment, the water molecules tend to aggregate around the hydrophobic regions or side chains of the protein, creating water shells of ordered water molecules. An ordering of water molecules around a hydrophobic region increases order in a system and therefore contributes a negative change in entropy (less entropy in the system). The water molecules are fixed in these water cages which drives the hydrophobic collapse, or the inward folding of the hydrophobic groups (Figure 2.38).

Figure 2.38 Formation of a Water Clathrate. Chloroform is a hydrophobic compound, thus, when it is dissolved in water forming a hydrate, the hydrophobic hydration is accompanied by a negative entropy change due to the increased order in the surrounding water and the positive heat capacity change, often causing a positive ΔG. Similar water cages can associate around hydrophobic protein residues prior to correct folding.
The hydrophobic collapse introduces entropy back to the system via the breaking of the water cages which frees the ordered water molecules. The multitude of hydrophobic groups interacting within the core of the globular folded protein contributes a significant amount to protein stability after folding, because of the vastly accumulated van der Waals forces (specifically London Dispersion forces). The hydrophobic effect exists as a driving force in thermodynamics only if there is the presence of an aqueous medium with an amphiphilic molecule containing a large hydrophobic region. The strength of hydrogen bonds depends on their environment; thus, H-bonds enveloped in a hydrophobic core contribute more than H-bonds exposed to the aqueous environment to the stability of the native state.
Chaperones
Molecular chaperones are a class of proteins that aid in the correct folding of other proteins in vivo (Figure 2.39). Chaperones exist in all cellular compartments and interact with the polypeptide chain in order to allow the native three-dimensional conformation of the protein to form; however, chaperones themselves are not included in the final structure of the protein they are assisting in. Chaperones may assist in folding even when the nascent polypeptide is being synthesized by the ribosome. Molecular chaperones operate by binding to stabilize an otherwise unstable structure of a protein in its folding pathway, but chaperones do not contain the necessary information to know the correct native structure of the protein they are aiding; rather, chaperones work by preventing incorrect folding conformations.

Figure 2.39 Top View of the GroES/GroEL Bacterial Chaperone Complex Model
Image by: Wikipedia
#/media/File:GroES-GroEL_top.png){kind=link}
In this way, chaperones do not actually increase the rate of individual steps involved in the folding pathway toward the native structure; instead, they work by reducing possible unwanted aggregations of the polypeptide chain that might otherwise slow down the search for the proper intermediate and they provide a more efficient pathway for the polypeptide chain to assume the correct conformations. Chaperones are not to be confused with folding catalysts, which actually do catalyze the otherwise slow steps in the folding pathway. Examples of folding catalysts are protein disulfide isomerases and peptidyl-prolyl isomerases that may be involved in formation of disulfide bonds or interconversion between cis and trans stereoisomers, respectively.
Chaperones are shown to be critical in the process of protein folding in vivo because they provide the protein with the aid needed to assume its proper alignments and conformations efficiently enough to become “biologically relevant”. This means that the polypeptide chain could theoretically fold into its native structure without the aid of chaperones, as demonstrated by protein folding experiments conducted in vitro; however, this process proves to be too inefficient or too slow to exist in biological systems; therefore, chaperones are necessary for protein folding in vivo. Along with its role in aiding native structure formation, chaperones are shown to be involved in various roles such as protein transport, degradation, and even allow denatured proteins exposed to certain external denaturant factors an opportunity to refold into their correct native structures.
Protein Denaturation
A fully denatured protein lacks both tertiary and secondary structure, however, the primary protein sequence remains intact and the protein exists as a random coil (Figure 2.39). Under certain conditions some proteins can refold; however, in many cases, denaturation is irreversible. Cells sometimes protect their proteins against the denaturing influence of heat with enzymes known as heat shock proteins (a type of chaperone), which assist other proteins both in folding and in remaining folded (Figure 2.40). Heat shock proteins are expressed in response to elevated temperatures or other stresses. Some proteins never fold in cells at all except with the assistance of chaperones which either isolate individual proteins so that their folding is not interrupted by interactions with other proteins or help to unfold misfolded proteins, allowing them to refold into the correct native structure. This function is crucial to prevent the risk of precipitation into insoluble amorphous aggregates.
Figure 2.40 Protein Denaturation. Figure (1) depicts the correctly folded intact protein. Step (2) applies heat to the system that is above the threshold of maintaining the intramolecular protein interactions. Step (3) shows the unfolded or denatured protein. Colored regions in the denatured protein correspond to the colored regions of the natively folded protein shown in (1).
Diagram provided by: Scurran15
#/media/File:Process_of_Denaturation.svg){kind=link}
The external factors involved in protein denaturation or disruption of the native state include temperature, external fields (electric, magnetic), molecular crowding, and even the limitation of space, which can have a big influence on the folding of proteins. High concentrations of solutes, extremes of pH, mechanical forces, and the presence of chemical denaturants can contribute to protein denaturation, as well. These individual factors are categorized together as stresses. Chaperones are shown to exist in increasing concentrations during times of cellular stress and help the proper folding of emerging proteins as well as denatured or misfolded ones.
Under some conditions proteins will not fold into their biochemically functional forms. Temperatures above or below the range that cells tend to live in will cause thermally unstable proteins to unfold or denature (this is why boiling makes an egg white turn opaque). Protein thermal stability is far from constant, however; for example, hyperthermophilic bacteria have been found that grow at temperatures as high as 122 °C, which of course requires that their full complement of vital proteins and protein assemblies be stable at that temperature or above.
Hydolysis
Hydrolysis is the breakdown of the primary protein sequence by the addition of water to reform the individual amino acids monomer units (Figure 2.41).
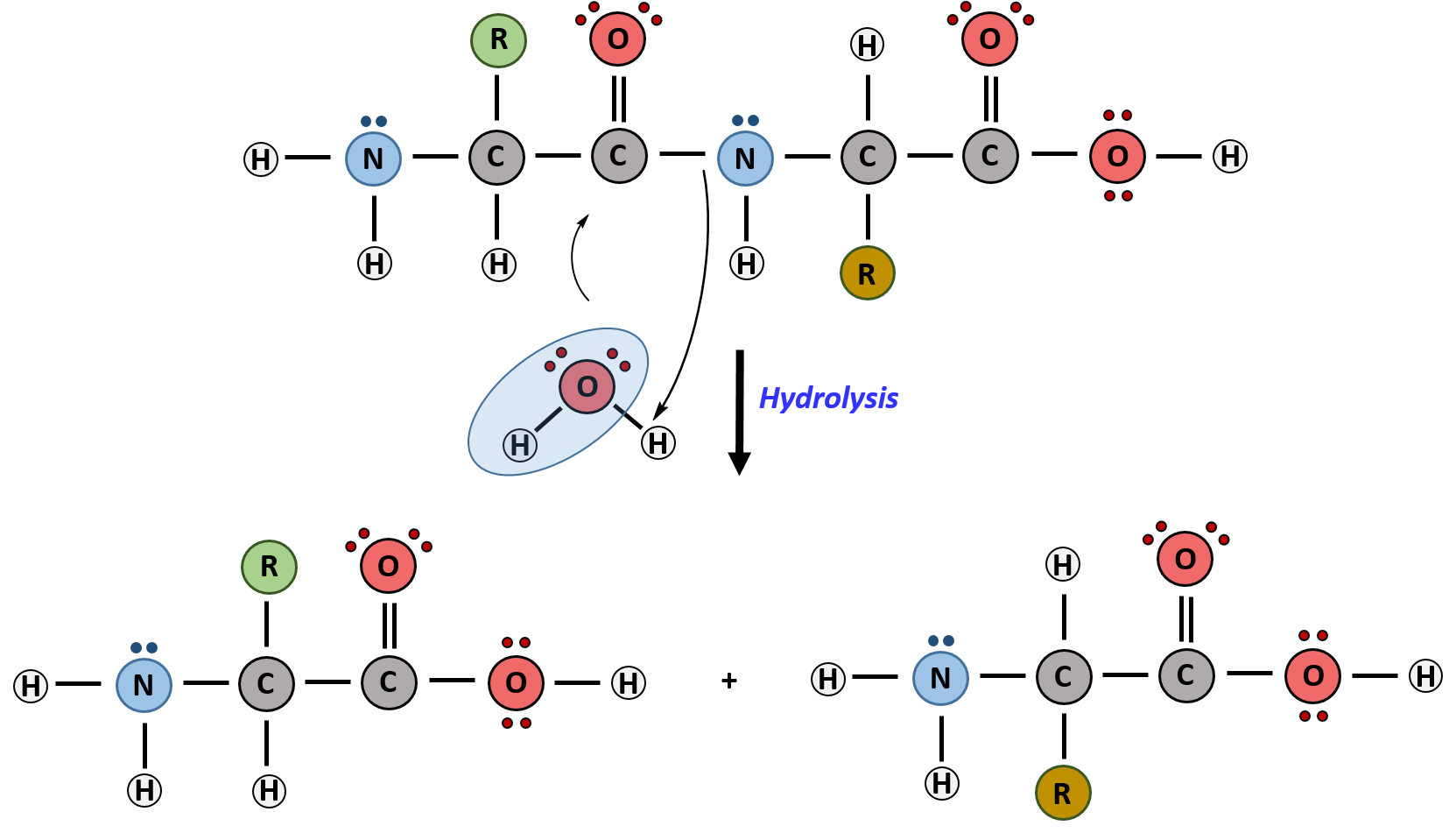
Figure 2.41 Hydrolysis of Proteins. In the hydrolysis reaction, water is added across the amide bond incorporating the -OH group with the carbonyl carbon and reforming the carboxylic acid. The hydrogen from the water reforms the amine.
Proteins are involved in many cellular functions. Proteins can act as enzymes which enhance the rate of chemical reactions. In fact, 99% of enzymatic reactions within a cell are mediated by proteins. Thus, they are integral in the processes of building up or breaking down of cellular components. Proteins can also act as structural scaffolding within the cell, helping to maintain cellular shape. Proteins can also be involved in cellular signaling and communication, as well as the transport of molecules from one location to another. Under extreme circumstances such as starvation, proteins can also be used as an energy source within the cell.
back to the top
2.7 References
OpenStax, Proteins. OpenStax CNX. Sep 30, 2016 http://cnx.org/contents/bf17f4df-605c-4388-88c2-25b0f000b0ed@2.
File:Chirality with hands.jpg. (2017, September 16). Wikimedia Commons, the free media repository. Retrieved 17:34, July 10, 2019 from https://commons.wikimedia.org/w/index.php?title=File:Chirality_with_hands.jpg&oldid=258750003.
{kind=link}
Wikipedia contributors. (2019, July 6). Zwitterion. In Wikipedia, The Free Encyclopedia. Retrieved 21:48, July 10, 2019, from https://en.wikipedia.org/w/index.php?title=Zwitterion&oldid=905089721
Wikipedia contributors. (2019, July 8). Absolute configuration. In Wikipedia, The Free Encyclopedia. Retrieved 15:28, July 14, 2019, from https://en.wikipedia.org/w/index.php?title=Absolute_configuration&oldid=905412423
Structural Biochemistry/Enzyme/Active Site. (2019, July 1). Wikibooks, The Free Textbook Project. Retrieved 16:55, July 16, 2019 from https://en.wikibooks.org/w/index.php?title=Structural_Biochemistry/Enzyme/Active_Site&oldid=3555410.
Structural Biochemistry/Proteins. (2019, March 24). Wikibooks, The Free Textbook Project. Retrieved 19:16, July 18, 2019 from https://en.wikibooks.org/w/index.php?title=Structural_Biochemistry/Proteins&oldid=3529061.
Fujiwara, K., Toda, H., and Ikeguchi, M. (2012) Dependence of a α-helical and β-sheet amino acid propensities on teh overall protein fold type. BMC Structural Biology 12:18. Available at: https://bmcstructbiol.biomedcentral.com/track/pdf/10.1186/1472-6807-12-18
Wikipedia contributors. (2019, July 16). Keratin. In Wikipedia, The Free Encyclopedia. Retrieved 17:50, July 19, 2019, from https://en.wikipedia.org/w/index.php?title=Keratin&oldid=906578340
Wikipedia contributors. (2019, July 13). Alpha-keratin. In Wikipedia, The Free Encyclopedia. Retrieved 18:17, July 19, 2019, from https://en.wikipedia.org/w/index.php?title=Alpha-keratin&oldid=906117410
Open Learning Initiative. (2019) Integumentary Levels of Organization. Carnegie Mellon University. In Anatomy & Physiology. Available at: https://oli.cmu.edu/jcourse/webui/syllabus/module.do?context=4348901580020ca6010f804da8baf7ba.
Wikipedia contributors. (2019, July 16). Collagen. In Wikipedia, The Free Encyclopedia. Retrieved 03:42, July 20, 2019, from https://en.wikipedia.org/w/index.php?title=Collagen&oldid=906509954
Wikipedia contributors. (2019, July 2). Rossmann fold. In Wikipedia, The Free Encyclopedia. Retrieved 16:01, July 20, 2019, from https://en.wikipedia.org/w/index.php?title=Rossmann_fold&oldid=904468788
Wikipedia contributors. (2019, May 30). TIM barrel. In Wikipedia, The Free Encyclopedia. Retrieved 16:46, July 20, 2019, from https://en.wikipedia.org/w/index.php?title=TIM_barrel&oldid=899459569
Wikipedia contributors. (2019, July 16). Protein folding. In Wikipedia, The Free Encyclopedia. Retrieved 18:30, July 20, 2019, from https://en.wikipedia.org/w/index.php?title=Protein_folding&oldid=906604145
Wikipedia contributors. (2019, June 11). Globular protein. In Wikipedia, The Free Encyclopedia. Retrieved 18:49, July 20, 2019, from https://en.wikipedia.org/w/index.php?title=Globular_protein&oldid=901360467
Wikipedia contributors. (2019, July 11). Intrinsically disordered proteins. In Wikipedia, The Free Encyclopedia. Retrieved 19:52, July 20, 2019, from https://en.wikipedia.org/w/index.php?title=Intrinsically_disordered_proteins&oldid=905782287