Home » Student Resources » Online Chemistry Textbooks » CH450 and CH451: Biochemistry - Defining Life at the Molecular Level » Chapter 11: Translation
MenuCH450 and CH451: Biochemistry - Defining Life at the Molecular Level
Chapter 11: Translation
11.1 Overview of Translation
11.2 Transfer RNA (tRNA) Structure
11.3 Aminoacyl tRNA Synthetases
11.4 Ribosome Structure
11.5 Initiation of Protein Translation
11.6 The Elongation Phase of Translation
11.7 Translation Termination
11.8 Regulation of Translation
11.9 References
11.1 Overview of Translation
Within this chapter, we will cover the details of prokaryotic and eukaryotic translation. Translation is the process of converting the information housed in mRNA into the protein sequence. Essentially, you are translating the language of nucleotides into the language of amino acids. Recall that prokaryotic and eukaryotic transcription and translation systems differ in large part due to the compartmentalization of larger eukaryotic cells. Due to this compartmentalization, transcription and translation are separated spatially and temporally within the cell. Transcription occurs within the nucleus of eukaryotes and translation occurs within the cytoplasm (Fig. 11.1 B). Prokaryotes do not have compartmentalization and have, thus, evolved a coupled trancription/translation system where both process occur simultaneously (Fig. 11.1 A).

Figure 11.1 Cellular Location of Transcription and Translation in Prokayotes and Eukaryotes. (a) Prokaryotes lack cellular compartmentalization and show coupled transcription-translation processing, whereas (b) eukaryotes have a high degree of compartmentalization and separate the processes of transcription, which is in the nucleus of the cell, from the processes of translation, which is localized in the cytoplasm.
Figure from: Baccei, A., and Rice, M. Lumen Learning
Recall that peptide formation is a dehydration reaction that combines the carboxylic acid of the upstream amino acid with the amine functional group of the downstream amino acid to form an amide linkage (Fig. 11.2). Water is the by-product. The ribosome (a large complex of peptides and rRNA molecules) serves as the enzyme that mediates this reaction. It requires a mature mRNA to serve as the template, and performs peptide bond synthesis in a directional fashion from the N to the C-terminal of the growing peptide/protein. This is known as N- to C-synthesis.
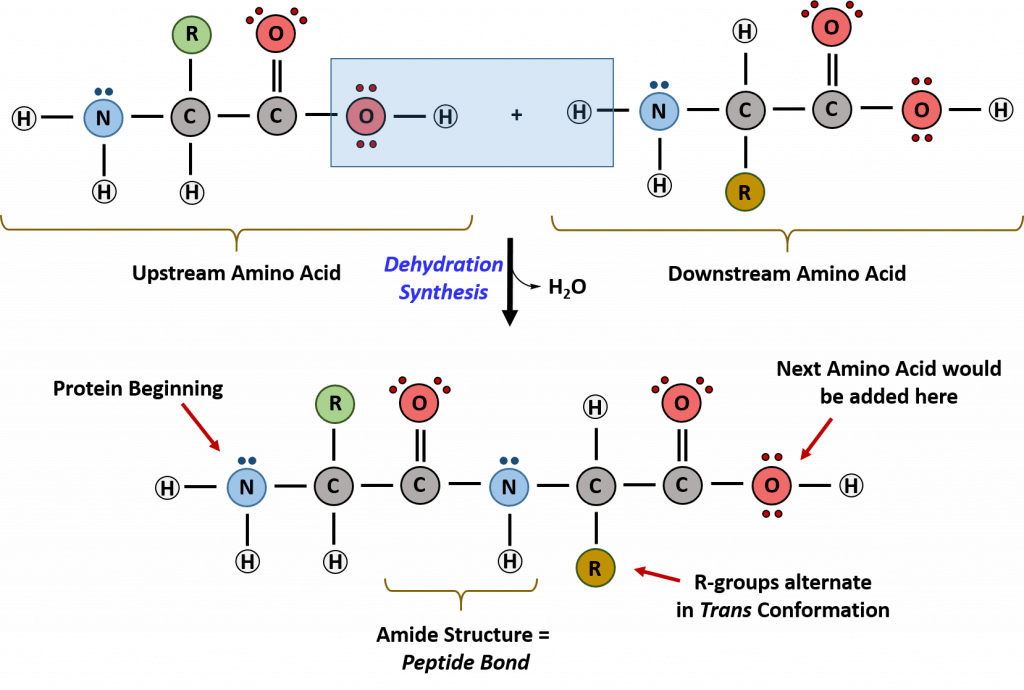
Figure 11.2 Formation of the Peptide Bond.The addition of two amino acids to form a peptide requires dehydration synthesis. The carboxylic acid of the upstream amino acid is joined with the amine functional group of the downstream amino acid to form the amide linkage. Within the ribosome, this reaction is highly directional and only occurs in the N to C orientation.
To maintain proper protein function, the error rate of translation is approximately 10-4 or 1 error in every 10,000 amino acids encoded. The fidelity of protein synthesis is maintained by the ribosomes ability to match the code from the template mRNA stand with the appropriate amino acid. Template mRNA is read by the ribosome in groups of three nucleotides, called a codon (Fig. 11.3 A). The template is non-overlapping and read in discrete groups of three. This is known as the reading frame of the mRNA, and it is always read from the 5′ to 3′ direction. Thus, for each mRNA there are three potential reading frames (Fig. 11.3 B). Only one reading frame will be the correct one for protein synthesis. The ribosome must recognize and align the correct reading frame of the mRNA such that the correct codon sequences can be read. Small distinct tRNA molecules are tethered with specific amino acids and contain specific anticodons that complement mRNA codon sequences. The tRNA molecules can cycle on and off of the ribosome structure to hybridize with the correct codon sequences and chaperone the correct amino acid for peptide bond formation. The ribosome then serves as a ribozyme and mediates the peptidyl transferase activity to form the peptide bond. The mRNA is then shifted to reveal the next codon within the sequence and the process is repeated until the entire protein has formed. Figure 11.3 C shows the codon chart for all of the possible combinations of three nucleotides. Note that there are 64 possible codon combinations using the 4 nucleotide possibilities, but only 20 amino acids that can be encoded for during protein synthesis. Each codon is specific for a single amino acid. There is very little ambiguity within the code.
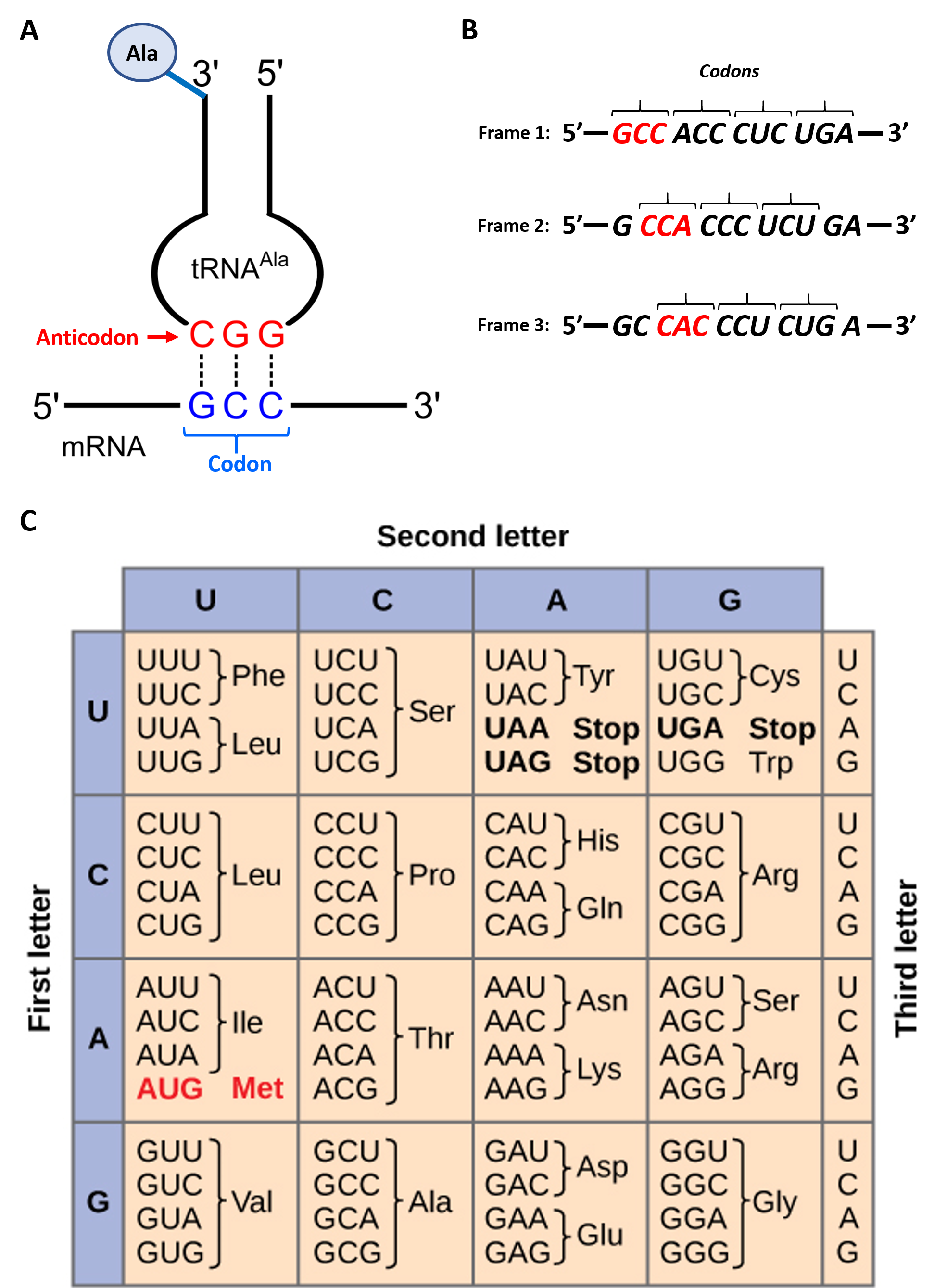
Figure 11.3 Reading the mRNA Template
Figure modified from: (A) Yikrazuul and (C) Zedalis, J., et al. Openstax
{kind=link}
However, there is redundancy within the code; i.e. many amino acids have more than one codon that encode for that specific amino acid. To account for this redundancy, many tRNA molecules can recognize more than one codon using a single anticodon. This is known as degeneracy. Degeneracy usually occurs at the 3rd position of the codon and is known as the wobble base position. Degeneracy helps to minimize the effects of mutations within the coding sequence, as mutations in the wobble base position will often lead to silent mutation– ie the mutation will still encode for the same amino acid.
In addition, if comparing the polarity of amino acids encoded by the different codons, neighboring codons typically encode for amino acids with similar polarity (Fig. 11.4). This also helps to minimize the effects of mutations, by converting one amino acid within the sequence to one that has similar polarity. This type of mutation is more likely cause less disturbance to the 3-dimensional structure of the resulting protein and retain biological function.

Figure 11.4 The Relationship of Codons and the Polarity of the Encoded Amino Acids. A representation of the genetic code, as function of the measured polarity of each codon. The smoothness of the landscape shows that moving from one AA to its neighbor does not change the polarity too abruptly.
Figure from: Eckmann, J-P., Rougemont, J., and Tlusty, T. (2019) ResearchGate
Degeneracy within the genetic code also allows for differential A/T & G/C concentrations within species. For example, the G/C content of bacteria can range from as low as 30% to as high as 70%. Organisms living at high temperatures or extreme environments often have higher G/C content. This effectively increases the hydrogen bond strength between the strands of the DNA (G/C pairs have 3 H-bonds, whereas A/T pairs only have 2) and causes an increase in the melting temperature of the chromosome. Thus, the DNA is stable at a higher temperature or under more extreme ionic conditions, such as high salt. A more detailed discussion of how a single tRNA can function to recognize more than one codon is the topic of the next section.
The Genetic Code is universal for almost all species alive on the planet, providing support for a single origin of life. Most deviations in the code occur within the mitochondria of eukaryotic species (Fig 11.5).
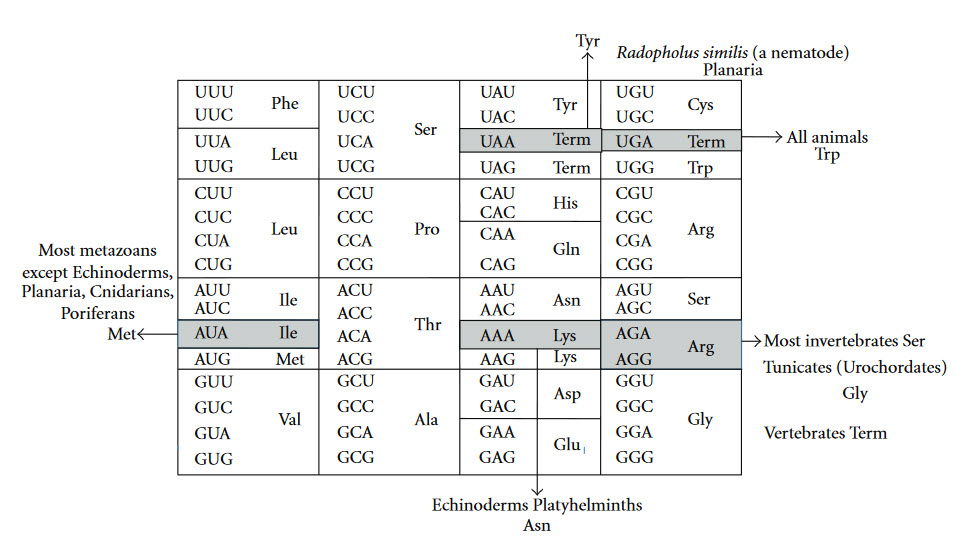
Figure 11.5 Variations in the Genetic Code. Universal genetic code (inside the box) and variations in animal mitochondrial genetic code (outside the box). Term = Termination Codon.
Figure from: Watanabe, K., and Yokobori, S. (2011) J. Nuc. Acids Article ID: 623095, 12 pages
Back to the Top
11.2 Transfer RNA (tRNA) Structure
Transfer RNAs (tRNAs) are central players in translation, functioning as adapter molecules between the informational level of nucleic acids and the functional level of proteins. Typically, tRNA molecules are between 76 – 90 nucleotides in length, and show a highly conserved secondary and tertiary structure. They also show the highest amount of nucleotide modification of all types of RNA with modifications concentrated in two hotspots—the anticodon loop and the tRNA core region, where the D- and T-loop interact with each other, stabilizing the overall structure of the molecule (Fig. 11.6 A). These modifications can cause large rearrangements as well as local fine-tuning in the 3D structure of a tRNA.
The life of a transfer RNA (tRNA) molecule starts with a series of important maturation steps that can vary in their sequential order from case to case. Leader and trailer sequences are removed by a set of endo- and exonucleases, and in several tRNA precursors, splicing reactions excise intronic sequences. Furthermore, in many organisms the sequence CCA, that represents the site of amino acid attachment, is not encoded, but has to be added post-transcriptionally by CCA-adding enzymes. While all primary tRNA transcripts are composed of the four standard RNA bases A, C, G and U, many of these nucleotides are modified, altering their properties in very different ways. Currently, 93 post-transcriptional modifications are known, and the variety of their functions is at least similarly diverse and not fully understood. The complexity of such modifications ranges from simple methylations at the bases or the ribose to rather complex and large base hypermodifications, whose synthesis often requires a whole cascade of enzymatic reactions. Modifications can alter a tRNA’s shape in subtle ways, but can also lead to massive structural rearrangements. In addition, they ensure efficient translation by maintaining the anticodon loop structure and promoting correct codon-anticodon interactions.
After maturation, tRNAs have multiple interaction partners in their life cycle, ranging from aminoacyl-tRNA-synthetases that are responsible for amino acid attachment, to translation factors, ribosomes and mRNAs. Apart from synthetases, these interaction partners do not specifically act on one individual tRNA transcript or isoacceptor, but on all tRNAs, similar to the above mentioned CCA-adding enzyme. Thus, despite a high sequence variation, a cell’s tRNAs show a well-conserved cloverleaf-like secondary structure that was originally discovered in 1965 (Fig. 11.6 A). The similar structure of all tRNA molecules allow them to bind to common protein synthesis machinery, such as the ribosome and CCA-adding enzymes. The cloverleaf consists of five parts: the acceptor stem (containing the tRNA’s 5′- and 3′-ends), the D-arm, the anticodon arm, the variable loop and the TΨC-arm (T-arm). At the 3′-terminus, the tRNA carries the CCA-sequence, required for aminoacylation, tRNA positioning in the ribosome and translation termination. In a conserved network of tertiary interactions, mostly between D- and T-loop, tRNAs fold into an L-shaped three-dimensional structure, which was first solved by Kim et al. in 1974 (Fig. 11.6 B). The anticodon and the amino acid-accepting CCA-end separated by the longest possible distance from each other. This conserved structure of a tRNA is essential for its recognition by the ribosome, other RNAs and proteins and, consequently, for its functionality. For example, the CCA-adding enzyme uses the acceptor domain for substrate recognition, whereas aminoacyl-tRNA-synthetases use several recognition elements like anticodon, acceptor stem or the discriminator position.
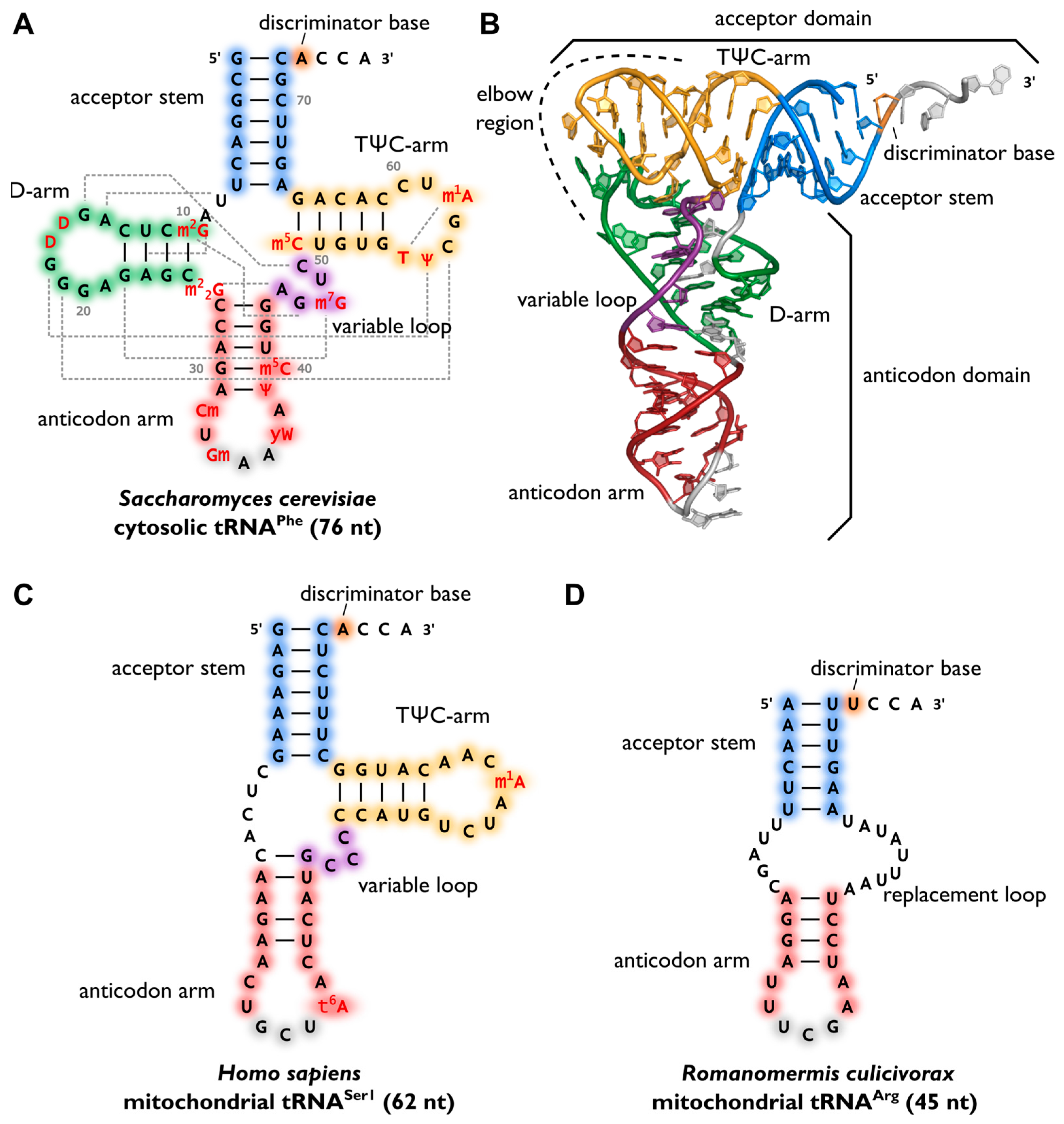
Figure 11.6.Variability of transfer RNA (tRNA) structures. (A) The canonical cloverleaf secondary structure of cytosolic tRNAPhe from S. cerevisiae is shown with acceptor stem (blue), D-arm (green), anticodon arm (red), variable loop (purple) and TΨC-arm (yellow). The anticodon is labeled in grey, the discriminator base in orange and post-transcriptional modifications in red. Grey dashed lines indicate tertiary interactions based on structural data and the length of the RNA is indicated in parenthesis; (B) The L-shaped tertiary structure of the cytosolic tRNAPhe from S. cerevisiae. Protein Data Bank entry (PDB): 1EHZ. The acceptor domain is composed of stacked T-arm and acceptor stem, whereas D- and anticodon arm form the anticodon domain. The region where both domains come together and interact with each other via tertiary base pairing is also called elbow region; (C) Secondary structure of human mitochondrial tRNASer1, which lacks the whole D-arm; (D) Secondary structure of the mitochondrial tRNAArg from the nematode Romanomermis culicivorax, which lacks both D- and T-arm. Instead, we find a so-called replacement loop. It represents the shortest tRNA found in vivo.
Figure from: Lorenz, C., et. al. (2017) Biomolecules 7(2):35
Surprisingly, not all tRNAs fold into the canonical cloverleaf structure. Especially many mitochondrial tRNAs are reduced in length and sometimes completely lack the D- or T-arm (Fig. 11.6 C). In mitochondria of nematodes, this situation is carried to an extreme, as tRNAs lacking one or even both arms seem to be the rule (Fig.11.6 D).
Post-transcriptional enzyme-catalyzed modification of tRNA occurs at a number of base and sugar positions and influences specific anticodon–codon interactions and regulates translation, its efficiency and fidelity. This phenomenon of nucleoside modification is most remarkable and results in a rich structural diversity of tRNA of which over 93 modifications have been characterized.
The variety of post-transcriptional modifications can be classified into two groups according to their complexity. The first group comprises the majority of modified bases, which have simple methylations at the ribose or base moiety that are usually introduced by a single enzymatic reaction. Simple modifications can be found at almost every position of the tRNA molecule with a high density in the tRNA core region, where tertiary interactions between D- and T-arm stabilize the three-dimensional fold (Fig. 11.7). The second group includes complex modifications, whose synthesis requires the sequential activity of several enzymes. Most often these hypermodified nucleosides are found in the anticodon of tRNAs, where they play a direct role in codon recognition and create what is known as the wobble base or wobble position.
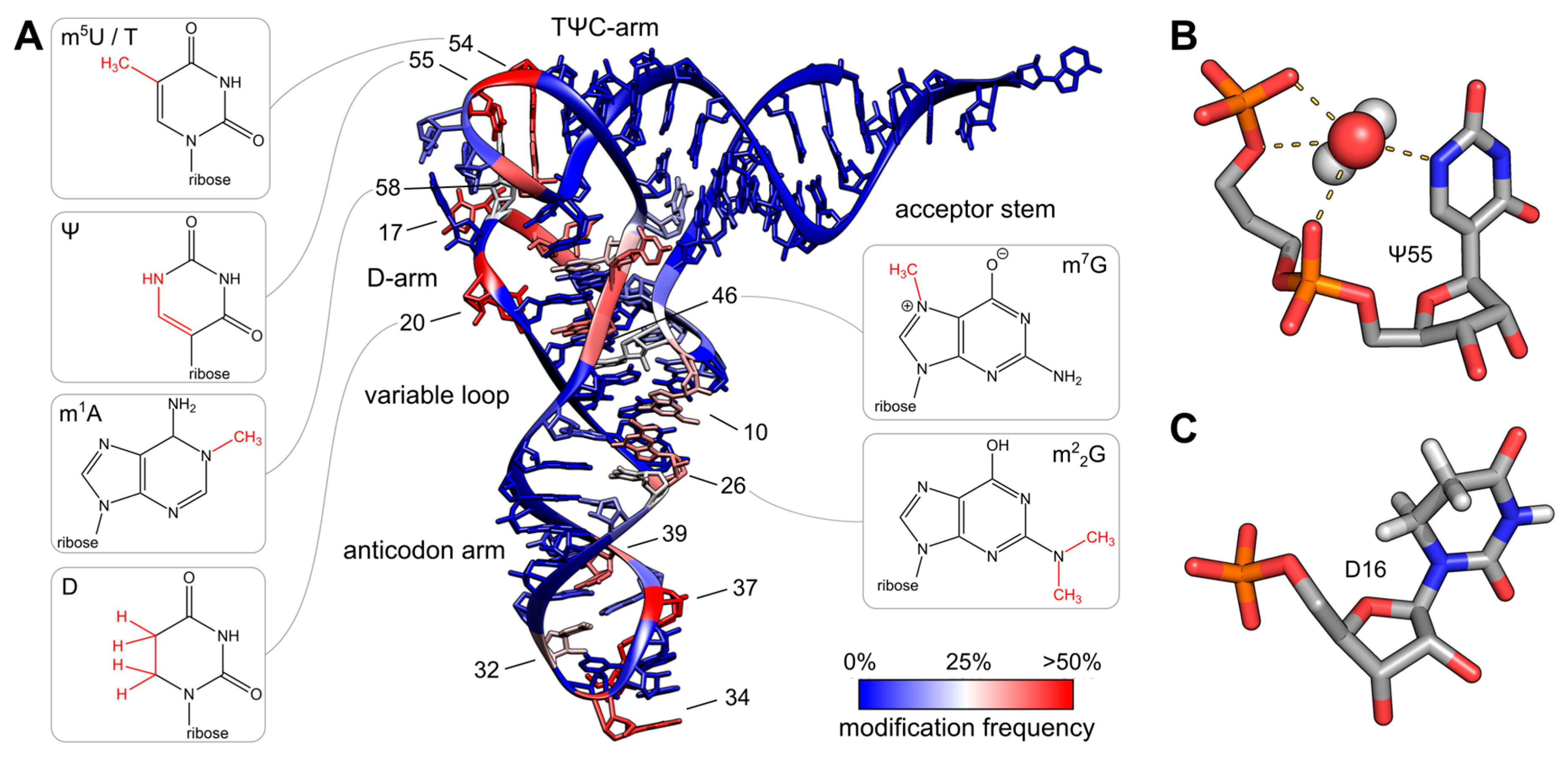
Figure 11.7 Post-transcriptional modifications in tRNA. (A) The colored tRNA structure shows the modification frequency of each base. The modification data were taken from the tRNAmodviz database and plotted on the crystal structure of tRNAPhe from S. cerevisiae. Blue colored bases are rarely modified; red colored bases are modification hotspots. tRNAs possess two regions with high modification levels—the anticodon loop (especially positions 34 and 37) and the core or elbow region, where D- and T-loop bases interact with each other and stabilize the tertiary fold. For some important positions, the chemical structure of the most frequent modification at this position is shown; (B) Three dimensional structure of pseudouridine at position 55 of tRNAPhe from S. cerevisiae. The additional H-bond donor at N1 interacts with the 5′-adjacent phosphates via a coordinated water molecule. The hydrogen bound to N1 was not resolved in the crystal structure. The ribose shows a stabilizing C3′-endo conformation. PDB: 1EHZ; (C) Three dimensional structure of D16 in the D-arm of tRNAiMet from Schizosaccharomyces pombe. The C5-C6 bond of dihydrouridine is reduced, which leads to a non-planar structure of the base. The ribose takes the less stable C2′ -endo conformation. PDB: 2MN0.
Figure from: Figure from: Lorenz, C., et. al. (2017) Biomolecules 7(2):35
A wobble base pairis a pairing between two nucleotides in RNA molecules that does not follow Watson-Crick base pair rules. The four main wobble base pairs are guanine-uracil (G-U), hypoxanthine-uracil (I-U), hypoxanthine-adenine (I-A), and hypoxanthine-cytosine (I-C) (Fig 11.8). In order to maintain consistency of nucleic acid nomenclature, “I” is used for hypoxanthine because hypoxanthine is the nucleobase of the inosine nucleotide; nomenclature otherwise follows the names of nucleobases and their corresponding nucleosides (e.g., “G” for both guanine and guanosine – as well as for deoxyguanosine). The thermodynamic stability of a wobble base pair is comparable to that of a Watson-Crick base pair. Wobble base pairs are fundamental in RNA secondary structure and are critical for the proper translation of the genetic code.

Figure 11.8 The Four Main Wobble Base Pairs. The top three diagrams represent the flexibility of the inosine nucleotide to form hydrogen bond interactions with cytosine, uracil, and adenine. tRNA and mRNA wobble base pairing also includes guanine hydrogen binding with uracil.
Figure from: Fdardel
{kind=link}
The wobble base position is usually the first position of the anticodon (read in the 5′ – 3′ direction), which aligns with the 3rd position of the mRNA codon. This helps to explain the degeneracy found within the genetic code (Figs. 11.3 & 11.9) Degeneracy means that a single tRNA is able to recognize multiple different codons within mRNA.
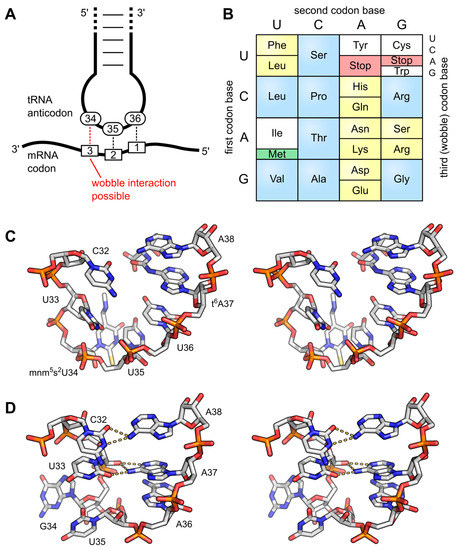
Figure 11.9 Anticodon Loop Structure and Codon Degeneracy. (A) The interaction of the anticodon bases (34–36) of a tRNA with the corresponding bases of the mRNA codons (3, 2, 1). A wobble interaction is possible between codon base 3 and anticodon base 34. The latter is frequently modified and directs the wobble interactions with the third codon base; (B) The standard genetic code is illustrated as a simple decoding table, 2-fold degenerate codon boxes are colored yellow, 4-fold degenerate boxes are blue. Start and stop codons are colored green and red, respectively; (C) Stereo image of the well-structured anticodon loop of tRNALys from E. coli. Modifications mnm5s2U34 and t6A37 prevent wrong base pairing inside the 7-nucleotide loop and promote the formation of the conserved U-turn motif. The stacked anticodon bases are located on the same side of the loop. PDB: 1FL8; (D) Stereo image of a collapsed and unmodified anticodon loop of tRNATyr from Bacillus subtilis. Here, bases 32 and 38 as well as 33 and 37 interact with each other and the U-turn motif is missing. The anticodon bases are not ordered and on opposite sides of the loop. PDB: 2LAC.
Figure from: Lorenz, C., et. al. (2017) Biomolecules 7(2):35
A prominent example is tRNAIle carrying the anticodon UAU. In principle, this anticodon can read codons AUA (for isoleucine) and AUG (for methionine). Yet, it was shown in some instances that tRNAIle with unmodified UAU anticodon exists, but has a strong preference for its cognate AUA codon, while it rarely misreads AUG. In most organisms, however, tRNAIle carries the anticodon CUA. To avoid misreading of the methionine codon by this tRNA, C34 (position 1 of the anticodon) is modified to lysidine (k2C34, chemical structure shown in Figure 11.10, which restricts codon recognition to only AUA and thereby changes the amino acid identity of the tRNA from methionine to isoleucine. In the archaeal species, Haloarcula marismortui, Methanococcus maripaludis and Sulfolobus solfataricus, this tRNAIle carries a different modification at C34, fulfilling the same purpose of restricting the interaction to AUA codons. Here, the original cytosine is modified at the C2-oxo position, which is replaced by agmatine (decarboxy-arginine), resulting in agmatidine (C+ or agm2C) (Fig 11.10). A complementary modification is that of N4-acetylcytosine (ac4C34, chemical structure shown in Figure 11.8) in elongator-tRNAMet of E. coli, which prevents the recognition of the AUA isoleucine codon. In non-plant mitochondria, however, both AUG and AUA codons are read as methionine. Hence, mitochondrial tRNAMet (carrying the anticodon CAU) has to recognize both codon forms. This is achieved by the introduction of 5-formylcytidine (f5C, Fig 11.10) at position 34, a modification that pairs with both A and U residues at the corresponding codon position 3.
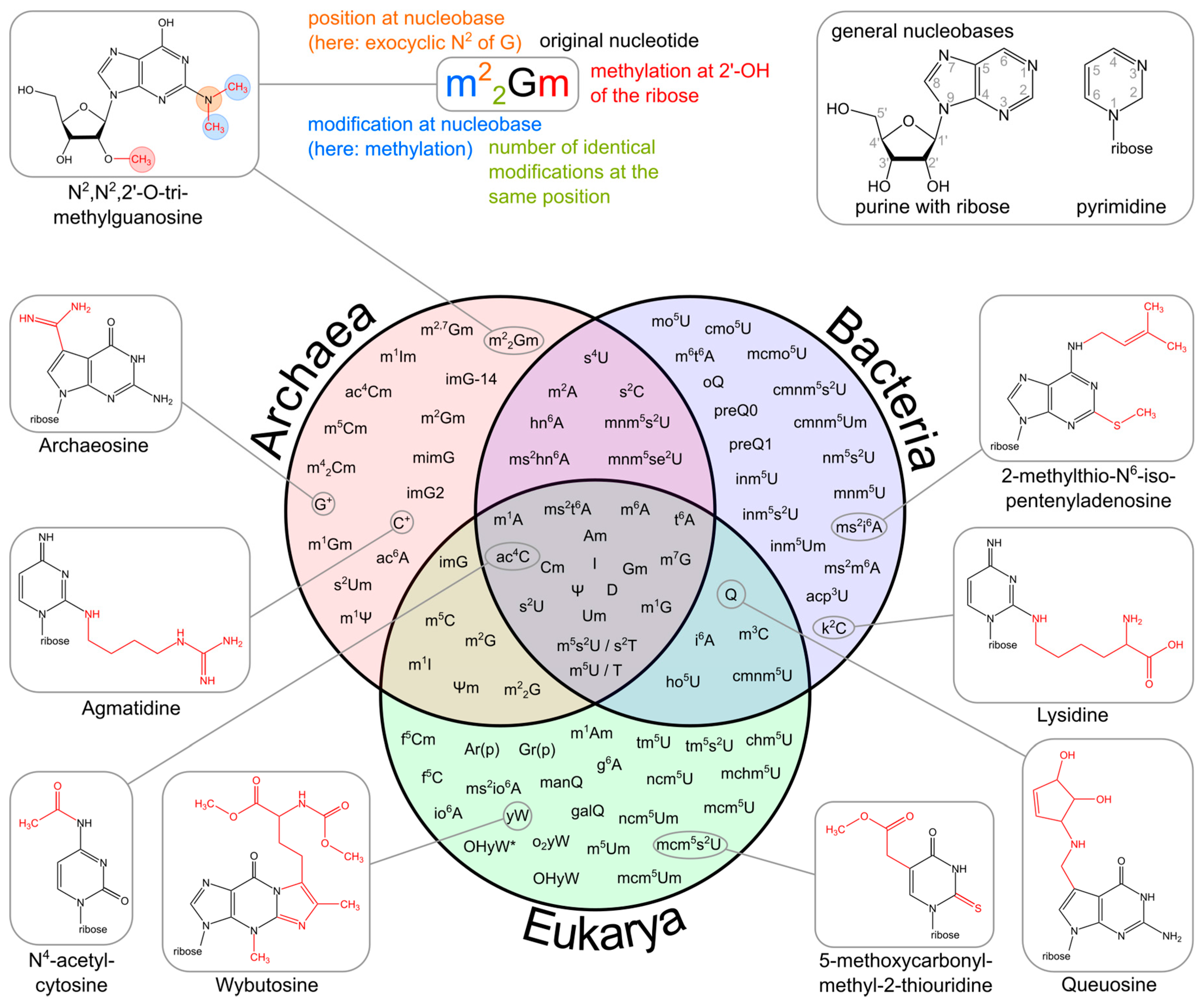
Figure 11.10 Variability of tRNA Modifications. The upper part of the image illustrates the systematic abbreviation of RNA modifications with N2,N2,2′-O-trimethylguanosine (m22Gm) as an example and also shows the atom numbering in the purine and pyrimidine rings as well as in the ribose. An abbreviation in front of the base letter describes a base modification, whereas letters after the base stand for ribose alterations. Superscripted numbers specify the position at the base and subscripted numbers indicate the frequency of identical modification at the same position. Abbreviations are as follows: ac—acetyl, acp—aminocarboxypropyl, chm—carboxyhydroxymethyl, cmo—oxyacetic acid, cmnm—carboxymethylaminomethyl, f—formyl, g—glycinyl, gal—galactosyl, hn—hydroxynorvalylcarbamoyl, ho—hydroxy, i—isopentenyl, inm—isopentenylaminomethyl, io—cis-hydroxyisopentenyl, m—methyl, man—mannosyl, mchm—carboxyhydroxymethyl methyl ester, mcm—methoxycarbonylmethyl, mcmo—oxyacetic acid methyl ester, mnm—methylaminomethyl, mo—methoxy, ncm—carbamoylmethyl, nm—aminomethyl, r(p) —5-O-phosphono-b-d-ribofuranosyl, s—thio, se—seleno, t—threonylcarbamoyl, tm—taurinomethyl. The Venn diagram summarizes data collected from the RNA modification database and contains the 93 post-transcriptional modifications that are found in tRNAs. Some examples mentioned throughout the text are shown with their chemical structure.
Figure from: Lorenz, C., et. al. (2017) Biomolecules 7(2):35
Organisms vary in the number of tRNA genes in their genome. For example, the nematode worm C. elegans, a commonly used model organism in genetics studies, has 29,647 genes in its nuclear genome, of which 620 code for tRNA.The budding yeast Saccharomyces cerevisiae has 275 tRNA genes in its genome.
In the human genome has approximately 20,848 protein coding genes, of which there are 497 nuclear genes encoding cytoplasmic tRNA molecules, and 324 tRNA-derived pseudogenes (tRNA genes thought to be no longer functional). Regions in nuclear chromosomes, very similar in sequence to mitochondrial tRNA genes, have also been identified (tRNA-lookalikes). These tRNA-lookalikes are also considered part of the nuclear mitochondrial DNA (genes transferred from the mitochondria to the nucleus).
As with all eukaryotes, there are 22 mitochondrial tRNA genes in humans. Mutations in some of these genes have been associated with severe diseases like the MELAS syndrome.
Cytoplasmic tRNA genes can be grouped into 49 families according to their anticodon features. These genes are found on all chromosomes, except the 22 and Y chromosome. High clustering on 6p is observed (140 tRNA genes), as well on 1 chromosome. Currently, it is unclear why their is so much redundancy within the genome to decode 61 of the 64 possible codons (the other three are stop codons used to terminate translation).
Back to the Top
11.3 Aminoacyl tRNA Synthetases
Aminoacyl-tRNA synthetases (aaRSs) are universally distributed enzymes that catalyze the esterification of a tRNA to its cognate amino acid (i.e., the amino acid corresponding to the anticodon triplet of the tRNA according to the genetic code). The product of this reaction, an aminoacyl-tRNA (aa-tRNA), is delivered by elongation factors to the ribosome to take part in protein synthesis.
Aminoacyl-tRNA synthetases are named after the aminoacyl-tRNA product generated, as such, methionyl-tRNA synthetase (abbreviated as MetRS) charges tRNAMet with methionine. In eukaryotes, an alternative nomenclature is often employed using the one-letter code of the amino acid (MARS) and a number is added to refer to the cytosolic (MARS1) or the mitochondrial (MARS2) variants. A total of 23 aaRSs have been described so far, one for each of the 20 proteinogenic amino acids (except for lysine, for which there are two) plus pyrrolysyl-tRNA synthetase (PylRS) and phosphoseryl-tRNA synthetase (SepRS), enzymes with a more restricted distribution that are only found in some bacterial and archaeal genomes. It is also worth noting that in eukaryotes the protein synthesis machineries of mitochondria and chloroplasts generally utilize their own, bacterial-like sets of synthetases and tRNAs that are distinct from their cytosolic counterparts.
The aminoacyl-tRNA synthetases catalyze a two-step reaction that leads to the esterification of an amino acid to the 3’ end of a tRNA along with the hydrolysis of one molecule of ATP, yielding aminoacyl-tRNA, AMP and PPi. In the first step, termed amino acid activation, both the amino acid and ATP bind to the catalytic site of the enzyme, triggering a nucleophilic attack of the α-carboxylate oxygen of the amino acid to the α-phosphate group of the ATP, condensing into aminoacyl-adenylate (aa-AMP), which remains bound to the enzyme, and PPi, which is expelled from the active site (Figure 11.11 A).
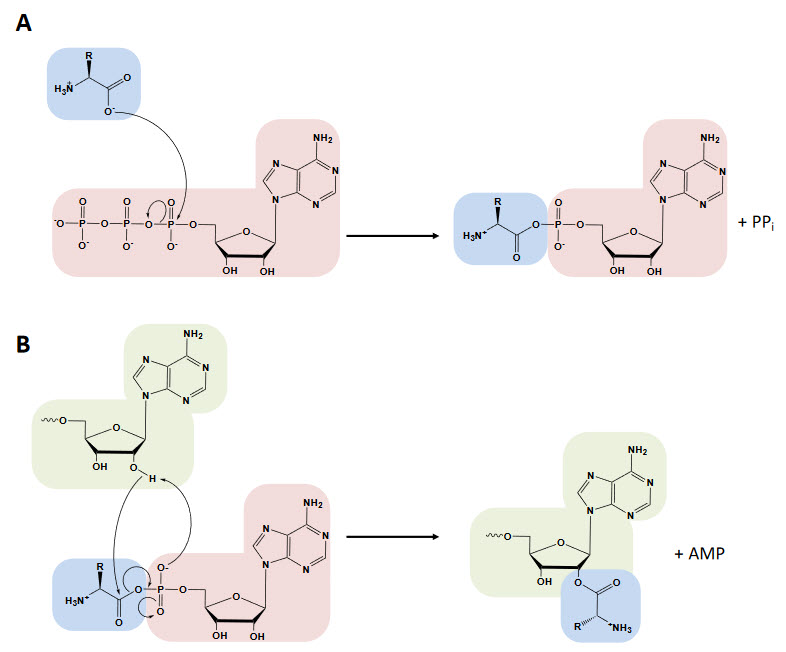
Figure 11.11 The Aminoacylation Reaction. In the first step, the amino acid (blue) is activated with ATP (red) in the synthetase active site (not depicted), forming the aminoacyl-AMP and releasing PPi. The amino acid is transferred to the tRNA (green) and AMP is released (depicted in the image transfer to the 3’-OH characteristic of class I aaRS, while in class II transfer happens at the 2’-OH with a 3’-OH attack in the second step).
Figure from: Gomez, M.A.R., and Ibba M. (2020) RNA,
Although tRNA is usually not required for this first step, certain synthetases do require the tRNA species for productive amino acid activation. In the second part of the reaction, either the 2′- or 3′-hydroxyl group of the terminal adenine nucleotide attacks the carbonyl carbon of the adenylate, forming aminoacyl-tRNA and AMP (Figure 11.12 B). While the two-step aminoacylation reaction is universally conserved, the aaRSs that catalyze it show extensive structural, and in some instances functional, diversity.
The 23 known aaRSs can be divided into two major classes based on the architecture of their active sites (Class I and Class II). In class I synthetases, the catalytic domain bears a dinucleotide or Rossman fold (RF) featuring a five-stranded parallel β-sheet connected by α-helices and is usually located at or near the N-terminus of the protein. This RF contains the highly conserved motifs HIGH and KMSKS, separated by a connecting domain termed connective peptide 1 (CP1) (Fig. 11.12 A). Class II active site architecture is organized as seven-stranded β-sheets flanked by α-helices and features three motifs which show a lesser degree of conservation than those in class I (Fig 11.12 B). Both classes also exhibit pronounced differences in their modes of substrate binding. Class I aaRSs bind the minor groove of the tRNA acceptor stem (with the exceptions of TrpRS and TyrRS) and aminoacylate the 2’-OH of the ribose of the terminal adenosine, while class II approach tRNA from the major groove and transfer amino acid to the 3’-OH (with the exception of PheRS). The mode of ATP binding is also different between both classes, being bound in an extended configuration in class I, while class II binds a bent configuration with the γ-phosphate folding back over the adenine ring. The kinetics of the aminoacylation reaction can also be used as a distinctive mechanistic signature, as aminoacyl-tRNA release is the rate limiting step for class I enzymes (except for IleRS and some GluRS) while for class II it is the amino acid activation rate instead.
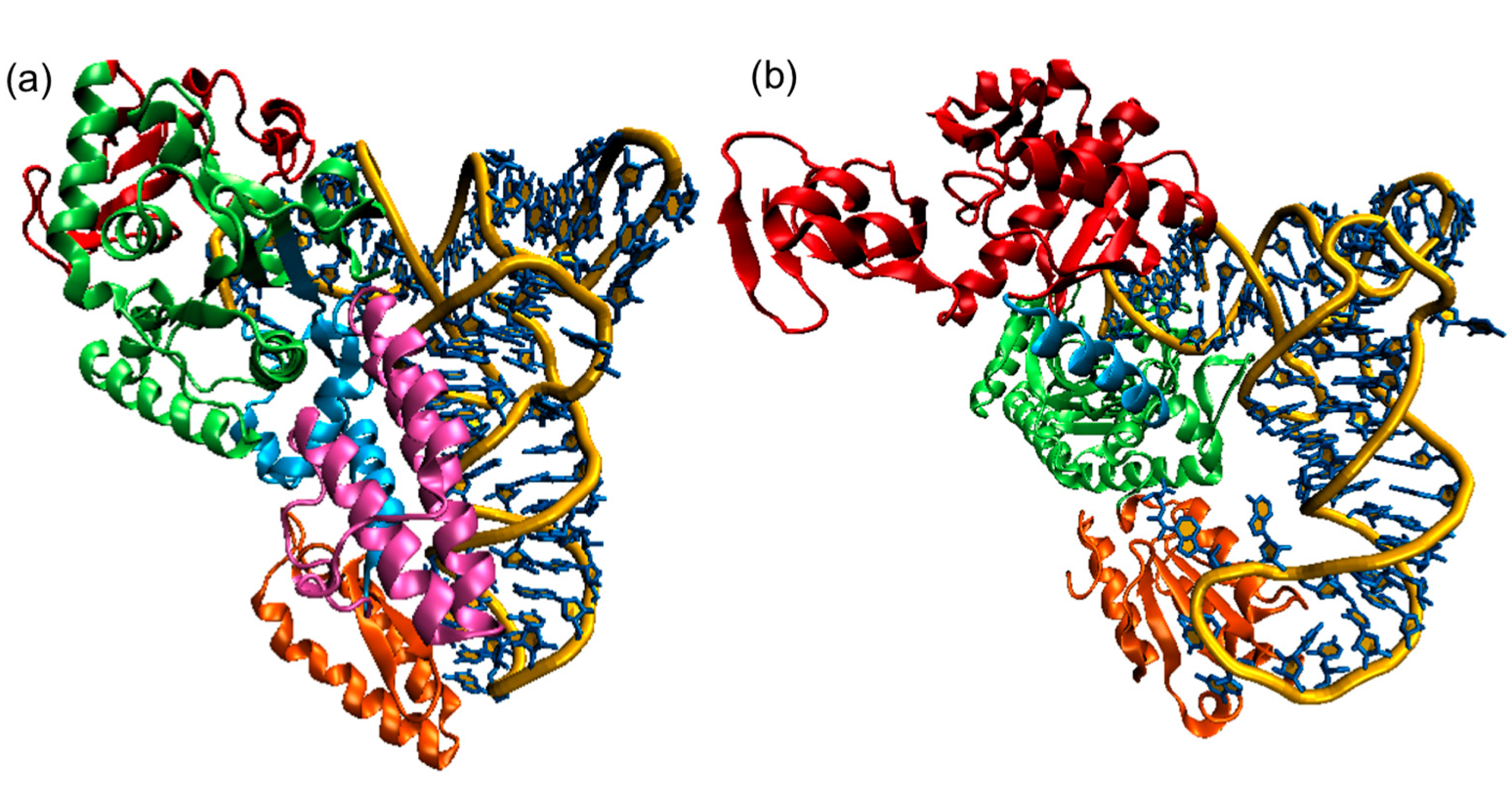
Figure 11.12 Structures of Class I and Class II AARSs (a) E. coliCysRS:tRNACys complex. The CP domain (red) and Rossmann fold catalytic domain (green), stem contact fold (cyan), helical bundle domain (magenta), and anticodon binding domain (orange) of CysRS are shown in a ribbon diagram; (b) A single monomer of the homodimeric E. coli ThrRS:tRNAThr complex. The two N-terminal domains (red), catalytic domain (green), linker (cyan), and anticodon binding domain (orange) of ThrRS are shown in a ribbon diagram. For both structures, the tRNAs are shown in a stick diagram (blue) with a trace of its backbone (yellow).
Figure from: Li, R., et. al. (2015) Int J. Mol. Sci. 16(7):15872-15902.
In order to ensure the faithful translation of the genetic message, synthetases must identify and pair particular tRNAs with their cognate amino acid which relies on the proper recognition of both substrates. This can prove extremely challenging for the synthetases as not only have they to discriminate the correct tRNA isoacceptor amongst a set of other tRNAs very similar in structure and chemical composition but also be able to select the cognate amino acid amidst an extremely large pool of similar amino acids, both proteinogenic and non-proteinogenic. The evolutionary pressure to maintain fidelity has driven aaRSs to develop an elevated specificity for their substrates, both the tRNA and the amino acid.
In addition, some synthetases have evolved editing activities that specifically target and hydrolyze misactivaed amino acids and/or misacylated tRNAs. To date, editing activity has been described in 10 out of the 23 aaRSs. In class I synthetases, this activity is located in the highly conserved CP1 domain, although in some enzymes such as MetRS and LysRS the editing activity resides in the catalytic site. In class II synthetases, however, the editing domains are more idiosyncratic. Editing mechanisms can be divided in two categories: pre- or post-transfer editing, in regard to the editing taking place before or after the transfer of the amino acid to the tRNA (Fig. 11.13).
Pre-transfer editing has been described in both class I and class II aaRSs and takes place after aa-AMP synthesis but before the aminoacyl moiety is transferred to the tRNA. Although the tRNA does not participate in the reaction itself, it has been reported that tRNA binding promotes editing activity in some aaRSs and is a requirement in IleRS and LeuRS. Pre-transfer editing can follow two main pathways. The first one is the selective release of the aa-AMP to the cytosol, where the labile phosphoesther bond is spontaneously hydrolyzed. The second route involves the enzymatic breakdown of the product and may happen either in the active site or in an independent editing site.
Post-transfer editing takes place after the transfer of the amino acid to the tRNA and involves the hydrolysis of the ester bond, in a domain separated from the active site. The specific mechanism of editing is idiosyncratic to each synthetase but in general, once formed the aa-tRNA triggers a conformational change and the 3’ terminus containing the aa is translocated from the active site to the editing site, sometimes traversing distances as large as 40 Å. As the core of the tRNA remains bound to the enzyme, this translocation often involves a rearrangement of the 3’ terminus to relocate to the editing site.
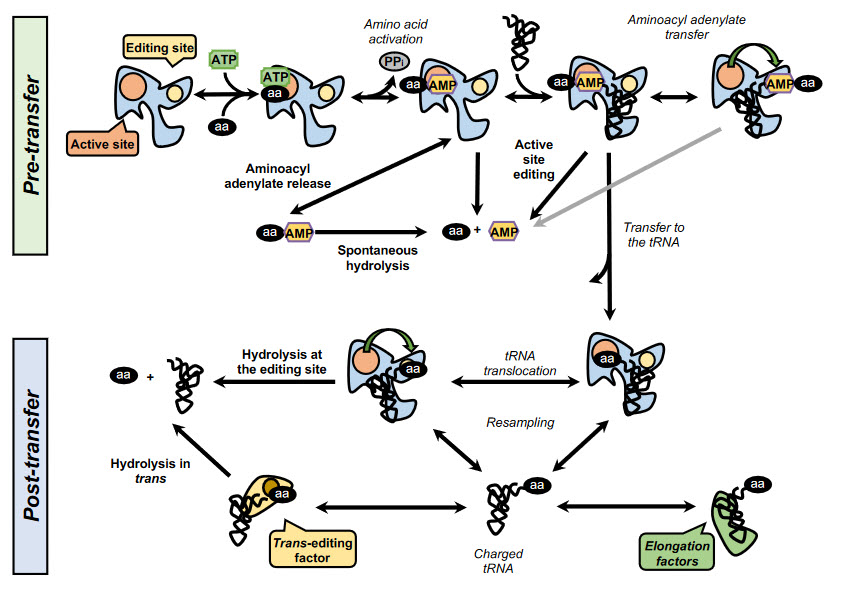
Figure 11.13 Aminoacyl tRNA Synthetase Editing Pathways. Schematic overview of the editing pathways employed by the synthetases. In the figure above, the events are in italics, while the editing paths are in bold. The pathways are divided between pre-transfer and post-transfer pathways. In the pre-transfer editing, the activated non-cognate aminoacyl-adenylate may be released from the enzyme and hydrolyze spontaneously or edited within the active site or a specialized active site. During post-transfer editing, the aminoacyl-tRNA can be translocated to the editing site or released and cleared by a dedicated trans-editing factor. When a correct cognate has formed, the resulting aminoacyl-tRNA binds the elongation factors and proceeds to translation in the ribosome.
Figure from: Gomez, M.A.R., and Ibba M. (2020) RNA,
Back to the Top
11.4 Ribosome Structure
The ribosome is a highly conserved molecular machine. In all organisms it is composed of two unequal subunits, which consist of a distinct set of ribosomal RNA (rRNA) and ribosomal proteins (RPs) that combine to form a large nucleoprotein complex. The ribosome structures in all living organisms, harbor three different tRNA binding sites: The A-site, where decoding occurs and the correct aminoacyl-tRNA (aa-tRNA) is selected on the basis of the mRNA codon displayed, the P-site, which carries the peptidyl-tRNA, and the E-site, which binds exclusively deacetylated tRNAs that are exiting the ribosome . Thus, during translation the tRNA moves from the A-site through the P- and E-site, where it leaves the ribosome (Fig. 11.14).
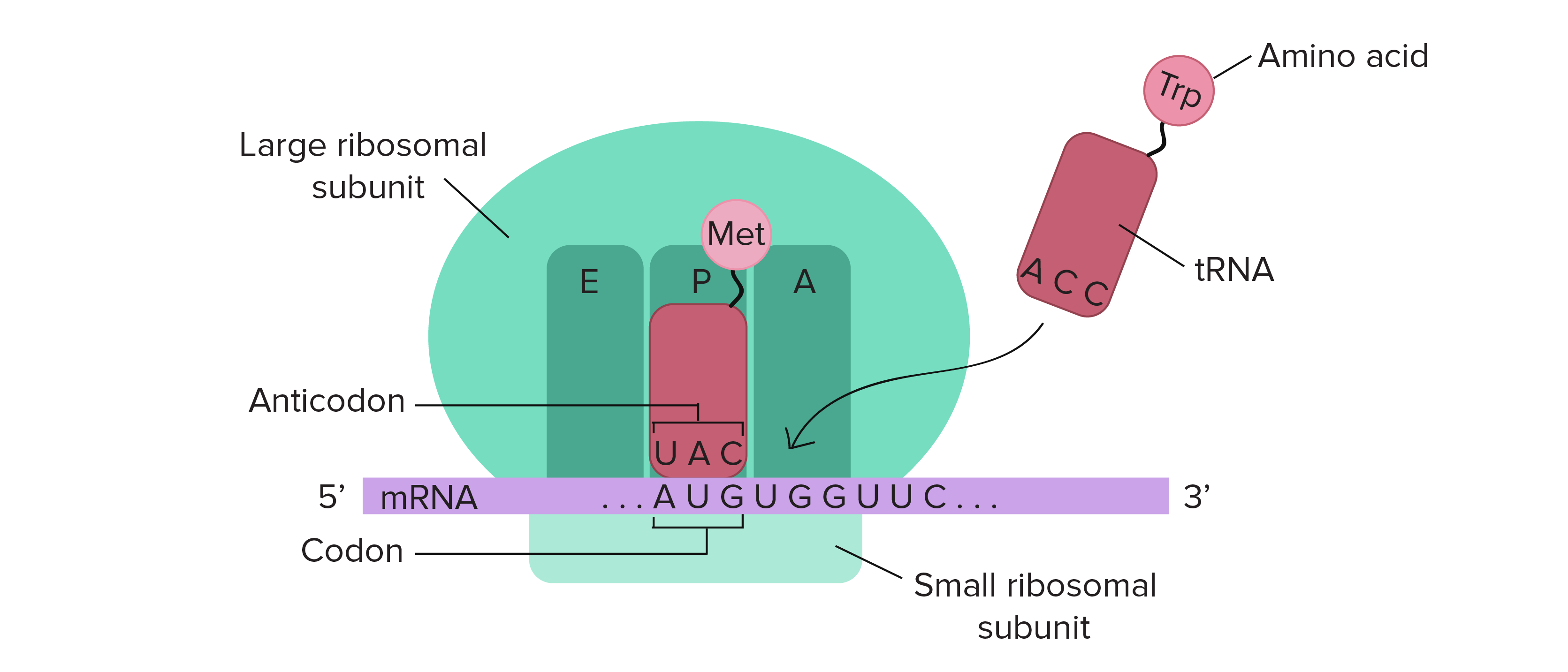
11.14 Schematic Structure of an Active Ribosome. The mRNA (shown in purple) is assembled between the small subunit and the large subunit of the ribosome (shown is green). tRNA molecules (shown in red) that are loaded with their cognate amino acid (shown in pink) are transitioned through the A-P-E sites of the ribosome during the elongation phase of translation. Movement of the tRNA molecules also shifts the position of the mRNA causing the next three codon bases to line up in the A-site of the ribosome.
Figure from: The Kahn Academy where it was modified from Openstax College Biology
The catalytic peptidyl transferase activity occurs when the tRNA molecules are bound in the A- and P-sites, transferring the nascent peptide to the incoming tRNA molecule (Fig. 11.15). Ribosomes are ribozymes, because the catalytic peptidyl transferase activity that links amino acids together is performed by the rRNA. Conceptionally, the complexity of the ribosome structure is reflected in the process of protein synthesis, which can be intersected into three major steps: initiation, elongation and termination/recycling.
Ribosomes are either free floating in the cytoplasm or they can be associated with the intracellular membranes that make up the rough endoplasmic reticulum (rER). Proteins translated into the rER will often be transported out of the cell or embedded into the plasma membrane (Fig 11.15).
Figure 11.15: A ribosome translating a protein that is secreted into the endoplasmic reticulum.
Figure from: Bensaccount
{kind=link}
Ribosomes from bacteria, archaea and eukaryotes in the three-domain system resemble each other to a remarkable degree. They differ in their size, much of the rRNA sequence, and the ratio of protein to RNA. Figure 11.16 shows the eukaryotic rRNA from the large subunit of the ribosome with highly conserved nucleotide elements (>90% sequence identity) within all of the domains of life, termed universal CNEs or uCNEs indicated. The differences in sequence and structure between eukaryotes and prokaryotes allow some antibiotics to kill bacteria by inhibiting their ribosomes, while leaving human ribosomes unaffected.

Figure 11.16 Universally Conserved Nucleotide Elements (uCNEs) in the Large Subunit of the Eukaryotic Ribosome.rRNA from the yeast large subuinit of the ribosome is shown from the crown view (from the subunit interface) with the L1 stalk at the upper left. uCNEs that are ≥90% conserved in sequence in all domains of life are indicated in dark blue.
Figure from: Doris, S.M., et. al. (2015) RNA 21:1719-1730
In all species, more than one ribosome may move along a single mRNA chain at one time (as a polysome), each “reading” its sequence and producing a corresponding protein molecule. In this way, many proteins can be translated from a single mRNA molecule. Within bacteria, translation is also coupled with transcription, as the two processes are not physically separated from one another. (Fig. 11.17). In eukaryotic organisms, polysomes form during translation. However, transcription and translation are not coupled, as the processes are separated into the nucleus and cytoplasm, respectively.
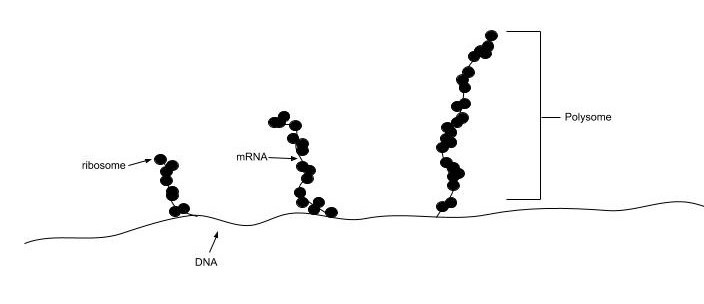
Figure 11.17 Bacterial Transcription and Translation are Coupled. Translation of nascent mRNA in bacteria occurs prior to the release of the nascent mRNA from the RNA Polymerase and DNA template. Multiple ribosomes assemble on the nascent mRNA and begin the translation of multiple protein products from the newly formed mRNA template. The structure of the ribosomes assembled on the mRNA is referred to as a polysome.
Figure from: Gonzalezmg1
{kind=link}
The mitochondrial ribosomes of eukaryotic cells functionally resemble many features of those in bacteria, reflecting the likely evolutionary origin of mitochondria.
Prokaryotic Ribosome Structure
Prokaryotic ribosomes have a mass of about 2500 kDa and a size of 70S (or Svedberg units: A Svedberg unit is a measure of the sedimentation rate in centrifuge and thus is representative of size). A complete ribosome (70S) can be dissociated into large subunit (50S) and a small subunit (30S) (Fig 11.18). The small subunit is formed by the interactions of 21 different proteins and a 16S RNA molecule, whereas the large subunit contains 34 different proteins and two RNA molecules, a 23S and a 5S species.
The rate-limiting step in protein synthesis is the formation of the 70S initiation complex that will be discussed in detail within the next section.

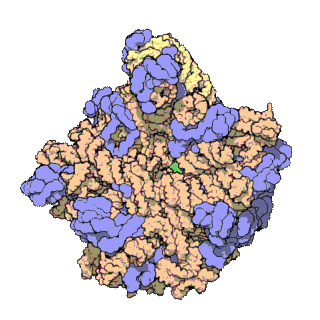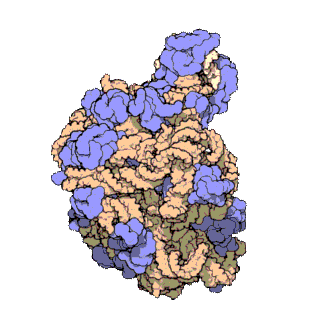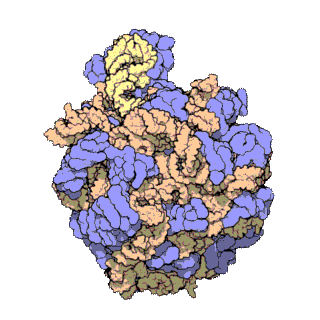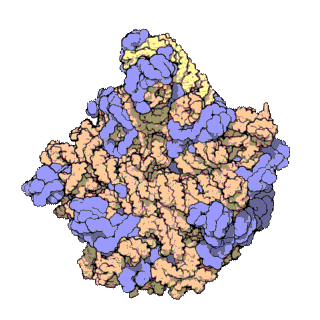
Figure 11.18 The Small and Large Prokaryotic Ribosomal Subunits(Left) Atomic structure of the 30S subunit from Thermus thermophilus.Proteins are shown in blue and the single RNA chain in orange. (Right) Atomic structure of the 50S subunit from Haloarcula marismortui. Proteins are shown in blue and the two RNA chains in orange and yellow. The small patch of green in the center of the subunit is the active site where the peptidyl transfer reaction occurs.
Figure from: Goodsell, D. (2000) Molecule of the Month
Eukaryotic Ribosome Structure
Eukaryotic ribosomes are larger than their prokaryotic counterparts at approximately 80S (although there is some modest variation between eukaryotic species). Human cytosolic ribosomes are composed of a large subunit (60S) that contains the 28S, 5.8S, and 5S rRNAs and 47 ribosomal proteins (RPs) and a small subunit (40S) that contains the18S rRNA and 33 RPs.
The assembly of eukaryotic ribosomal subunits starts in the nucleolus, where RNA polymerase I transcribes the major rRNA precursor (a 45S pre-RNA), from which, after processing and removal of the external and internal transcribed spacers (ETS and ITS), the mature rRNAs are generated (Figure 11.19). The pre-RNA is modified during transcription by small nucleolar ribonucleoproteins (snoRNPs), processed by RNA nucleases, and assembled with numerous RPs. After processing of the rRNA precursor, the pre-40S and pre-60S subunits follow separate biogenesis routes. Here we will describe the assembly of the 60S subunit in more detail.

Figure 11.19 Schematic Representation of Ribosomal RNA Processing in Eukaryotes. The 45S rRNA transcript is processed by endonucleotide cleavage and splicing to form the 5.8S and 28S components of the large ribosomal subunit and the 18S component of the small ribosomal subunit. The 5S subunit is transcribed independently from the other rRNA components and is incorporated into the large ribosomal subunit.
Figure from: FeatherPluma
{kind=link}
Although the exact assembly of the 60S subunit is not currently known, a model has been postulated that suggests that in the nucleolus, after circularization of rRNA domains, early 60S assembly is carried out in a sequential fashion (Fig. 11.20). As the transcription of the pre-rRNA proceeds, the rRNA quickly develops core secondary structure that promotes the interaction of key Assembly Factors (AFs) and RPs prior to trancriptional termination. Specifically during this time, the 5.8S portion, ITS2, and domains I and II and partially domain VI are folded in the earliest observed intermediate (state A in Fig. 11.20). Thus, it appears that the solvent-exposed back side of the large subunit forms like an exoskeleton and construction proceeds by formation of the exit tunnel. This model is in agreement with a previously suggested model of hierarchical folding based on RP depletion phenotypes. The peptidyl transferase center (PET) is predicted to be one of the later folding steps in the process (state F in Fig. 11.20) Although the very late-folding peptidyl transferase center is the evolutionary oldest part of the ribosome, it is likely that folding the solvent side first brings the advantage of providing a stable scaffold for the developing 60S subunit. The folding and assembly of the 40S subunit follows a similar pattern. The two subunits remain unattached until activated in the cytoplasm through the binding of a mRNA transcript with the small subunit. This begins the formation of the initiation complex that will mark the start of the transcriptional process.

Figure 11.20 Assembly Sequence of the Pre-rRNA Domains in the Large Subunit of a Eukaryotic Ribosome. Assembly of RPs and AFs to the nascent 35S rRNA precursor starts co-transcriptionally. Very early, the pre-rRNA is circularized as domain VI binds to domains I and II and the 5.8S portion of the precursor rRNA. The formation of the PET (displayed here as a black circle) starts with this circularization. Its maturation progresses as rRNA domains fold following this order: VI, V, III, and IV. Full assembly of the PET is only achieved when domain V is completely folded as observed in state F. After that, only few additional steps need to occur before the particles are exported to the cytoplasm, where they undergo final maturation.
Figure from: Kater, L., et. al. (2017) Cell 171(7):1599-1610
Back to the Top
11.5 Initiation of Protein Translation
Prokaryotic Initiation
The small subunit of the ribosome (30S) interprets the genetic information by selecting aminoacyl-tRNAs cognate to the mRNA codons in the decoding center. The large subunit (50S) carries the catalytic peptidyl transferase center where amino acids are polymerized into a protein. Small and large subunits unite together at the start codon of a gene to form the 70S ribosome and dissociate again at the stop codon upon completing the synthesis of the encoded protein. This process consists of three phases: initiation, elongation, and termination. In this section, we will focus on the initiation of translation.
In bacteria, the initiation phase of protein synthesis involves a limited number of “actors”. Aside from the two ribosomal subunits, key roles are played by the initiator tRNAfmet, the translation initiation region (TIR) of the mRNA, and three protein factors – the initiation factors (IFs) IF1, IF2 and IF3 – that ensure speed and accuracy to the overall process. The initiator tRNAfmet contains a methionine residue that has been enzymatically modified to contain an N-terminal formyl group (Fig. 11.21) fMet is only used for the initiation of protein synthesis and is thus found only at the N-terminus of the protein. Unmodified methionine is used during the rest translation. Once protein synthesis is completed, the formyl group on methionine may be removed by peptide deformylase, and on occassion, the entire methionine residue can be further removed by the enzyme methionine aminopeptidase.
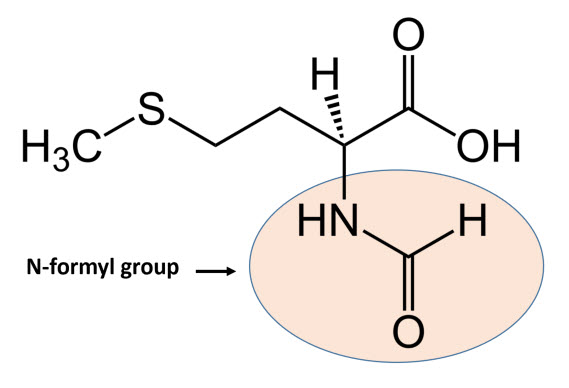
Figure 11.21 Structure of N-formylmethionine.
The TIR sequence within the mRNA contains the start codon and usually an upstream untranslated region that interacts with the small subunit of the ribosome. The bacterial cell produces and expresses a plethora of different mRNAs with different TIR sequences and structures; the efficiency by which these individual transcripts are translated depends not only upon their abundance and stability but also upon the nature of TIR. Thus, unlike the other aforementioned actors that represent constants, the mRNA TIRs represent essentially the only variable in the process of mRNA initiation site selection and can effect translation efficiency.
Although the triplet AUG is by far the most frequent initiation codon found in TIRs, other initiation triplets (i.e., GUG, UUG, AUU, AUC, and AUA) are found in bacteria and the central U is the only universally conserved base of the start codon. Among the aforementioned triplets, those having a 3′-G (i.e., AUG, GUG and UUG) are recognized equivalently and most efficiently by IF3 during the initiation complex formation.
Another important characteristic of a large number of bacterial mRNA TIRs is the presence of a Shine–Dalgarno (or SD) sequence that is complementary to the 3′ end sequence of 16S rRNA (the anti-SD sequence or aSD). The SD sequence, when present, is usually at an optimal distance of 4–9 nucleotides upstream of the initiation codon (Fig. 11.22). While the SD sequence plays an important role in the efficient translation of many mRNA transcripts, it is not essential. Many other mRNA sequences fully lack an SD sequence, but are still efficiently transcribed. Thus, the SD sequence is only one example of TIR mechanisms that can play an important role in mRNA binding with the small subunit of the ribosome.

Figure 11.22 Shine-Dalgarno Translation Initiation Sequence. Prokaryotic mRNA sequences often share a highly conserved sequence upstream of the start codon known as the Shine-Dalgarno sequence. This consensus sequence is complimentary to the 3′-end of the 16S rRNA sequence in the small subunit of the ribosome. It is an important feature for the binding and docking of many mRNA molecules with the small ribosomal subunit during transcription initiation.
Figure modified from: Shakeistone
{kind=link}
The three protein initiation factors, IF1, IF2, and IF3, determine the kinetics and fidelity of the overall initiation process. The three IFs are bound, one copy each, to specific sites of the 30S subunit where they assist with the formation of the initiation complex and assembly of the 70S ribosome.
As noted above, the initiator tRNA is first aminoacylated with methionine whose α-NH2 group is eventually blocked by a specific formyl transferase (TMF) to produce a tRNAfmet molecule. This modification prevents interaction with the elongation factor EF-Tu (which we will see plays an important role in the elongation phase of translation, but not the initiation phase!) Blocking EF-Tu binding ensures instead the recognition and binding of tRNAfmet by initiation factor IF2, effectively docking it with the 30S subunit. Furthermore, tRNAfmet binds with high affinity to the ribosomal P-site, unlike all other aminoacyl-tRNAs that bind to the A-site in a ternary complex with EF-Tu and GTP (details will be presented in the next section). In the P-site, the initiator tRNA must be recognized as correct by the other initiation factors IF3 and IF1.
To form the 30S initiation complex, IF3 and IF2 are the first factors to bind to the 30S subunit forming an unstable 30S-IF3-IF2 complex (Fig. 11.23 a). The binding of IF1 causes a conformational change in the 30S subunit stabilizing the complex and allowing the recruitment of the tRNAfmet by IF2. Notably, IF1 binds in the A site of the 30S subunit, where it contacts ribosomal protein S12. Recruitment of the tRNAfmet can also stabilize the mRNA interactions with the 30S subunit through the formation of hydrogen bonds between the codon of the mRNA and the anticodon of the tRNAfmet. Note that the binding of mRNA to the 30S subunit is IF-independent and can take place at any time during the 30S assembly process. Two potential routes of mRNA association are shown in Figure 11.23 (a) where the mRNA is assembled either prior to or after tRNAfmet recruitment.

Figure 11.23 Initiation of Transcription. Step 1: a vacant 30S ribosomal subunit binds IF3 and IF2. Step 2: IF1 binds to the 30S subunit in the presence of both IF3 and IF2. Steps 3 and 3′: in the presence of all three factors tRNAfmet is recruited. Steps 4 and 4′:the mRNA is bound with different on and off rates depending on its TIR structure; mRNAs with strong secondary structures are bound more slowly than those having little or no secondary structure. Step 5:mRNAs containing secondary structures must be unfolded in a process that is facilitated by IF2 bound to GTP and antagonized by IF3. Step 6:the isomerization of the 30S pre-IC allows P-site codon–anticodon interaction to yield a more stable 30SIC from which mRNA and fMet-tRNA are more stably bound. Step 7: a 30SIC, containing IF1, IF2·GTP, IF3 and mRNA whose initiation triplet is P-site decoded by fMet-tRNA, is docked by a 50S subunit. Step 8: upon contact with the 50S subunit, the GTPase function of IF2 is activated and GTP is rapidly hydrolyzed leaving GDP+Pi bound to IF2. Step 9:this reversible conformational transition represents the last kinetic checkpoint of translation initiation fidelity by IF3 and IF1, as IF3 and IF1 dissociate from the complex. Step 10:The first-order isomerization of the IF2-GDP structure causes a shift in the ribosome structure that represents the rate-limiting step in 70SIC formation. Step 11: Pi is dissociated from IF2·GDP. Step 12: IF2 leaves the ribosome (or moves away from the A-site) clearing the way for EF-Tu binding. Step 13:the EF-Tu·GTP·aminoacyl-tRNA complex binds to the 70SIC and through a number of steps (not represented here) delivers to the ribosomal A-site the aminoacyl-tRNA encoded by the second mRNA codon. Step 14:the tRNAfMet bound in the P-site of the peptidyl transferase center donates its formyl-methionine to the A-site-bound aminoacyl-tRNA to yield the initiation dipeptide fMet-aa. Initiation is then complete and the elongation phase can begin.
Figure from: Gualerzi, C.O., and Pon, C.L. (2015) Cell Mol Life Sci. 72:4341-4367.
Following the recruitment of the mRNA and the tRNAfmet to the 30S initiation complex loaded with the IF2, IF3 and IF1 initiation factors, the 50S subunit is very rapidly docked to yield an initially unstable 70S initiation complex (Fig 11.23 b). It should be noted that the IF2 protein is a GTP hydrolase enzyme and, as such, binds with the cofactor GTP prior to the recruitment of the 50S subunit. Contact between the IF2 GTPase activating center with the 50S subunit causes the rapid hydrolysis of GTP to GDP + Pi.
The formation of the 70S complex causes the dissociation of the initiation factors. IF2 is the last factor to be dissociated, leaving the ribosome after having positioned tRNAfMet in the P-site of the 70S initiation complex. It must be placed in the correct orientation to facilitate peptide bond formation. GDP and Pi also dissociate from the complex with the removal of IF2. The elongation factor, EF-G is then free to chaperone the first tRNA into the A-site and the first peptide bond is formed (Step 13 of Fig. 11.23 b). This marks the beginning of the elongation phase of protein synthesis.
Eukaryotic Initiation
Eukaryotic translation initiation is more complex than prokaryotic systems and requires the actions of at least 11 eukaryotic initiation factors (eIFs), plus additional auxiliary factors (Table 11.1). We will not cover the action of all these eIFs in detail here, but rather focus a few key steps (Fig. 11.24).
Table 11.1 Comparison of Prokaryotic and Eukaryotic Translation Initiation Factors

First, the initiator tRNAi is recruited to the small ribosomal subunit (40S) to form a ternary complex with the GTP-bound eukaryotic initiation factor 2 (eIF2). Formation of this 43S pre-initiation complex is strongly enhanced by additional factors, such as eIF3. eIF3 also interacts with the eIF4F complex, which consists of three other initiation factors: eIF4A, eIF4E, and eIF4G. eIF4G is a scaffolding protein that directly associates with both eIF3 and the other two components. eIF4E is the 5′-cap-binding protein. Binding of the mRNA cap by eIF4E is often considered the rate-limiting step of cap-dependent initiation, and the concentration of eIF4E is a regulatory nexus of translational control. Certain viruses cleave a portion of eIF4G that binds eIF4E, thus preventing cap-dependent translation to hijack the host machinery in favor of the viral (cap-independent) messages. eIF4A is an ATP-dependent RNA helicase that aids the ribosome by resolving certain secondary structures formed along the mRNA transcript. The poly(A)-binding protein (PABP) also associates with the eIF4F complex via eIF4G, and binds the poly-A tail of most eukaryotic mRNA molecules. This protein has been implicated in playing a role in circularization of the mRNA during translation.This 43S preinitiation complex accompanied by the protein factors moves along the mRNA chain toward its 3′-end, in a process known as ‘scanning’, to reach the start codon (typically AUG). After recognition of the start codon, the large ribosomal subunit (60S) assembles to form the 80S initiation complex, which is ready for elongation. Alternatively, under distinct conditions or on certain transcripts internal initiation can occur in a cap-independent manner at so called internal ribosome entry sites (IRES).
![]()
Figure 11.24 Eukaryotic Translation Initiation. This is a simplified diagram of eukaryotic translation initiation detailing some of the eIFs involved in the process. eIF2 is critical for recruiting the initiation tRNAi to the 40S subunit. eIF3 enhances the activity of eIF2 and also promotes the binding of the 43S pre-initiation complex with the mRNA. eIF3 binds with the mRNA through the interaction of the eIF4 factors and causes the scanning of the pre-initiation complex down the mRNA to locate the start codon (usually AUG). Poly A Binding Proteins (PABPs) bind with the polyA tail sequence of the mRNA and also interact with the eIF4 factors causing the circularization of the mRNA.
Figure from: Eukaryotic Translation, Wikiwand
As seen in prokaryotic systems with the favored Shine Dalgarno sequence upstream of the start codon within the mRNA sequence, there are also preferred nucleotide sequences within the local vicinity of the start codon in eukaryotic mRNAs, as well. In eukaryotic mRNA, this is known as the Kozak sequence (Fig. 11.25). The sequence was originally defined as 5′-(gcc)gccRccAUGG-3 where:
- The underlined nucleotides indicate the translation start codon, coding for Methionine.
- upper-case letters indicate highly conserved bases, i.e. the ‘AUGG’ sequence is constant or rarely, if ever, changes.
- ‘R’ indicates that a purine (adenine or guanine) is always observed at this position (with adenine being more frequent according to Kozak rules)
- a lower-case letter denotes the most common base at a position where the base can nevertheless vary
- the sequence in parentheses (gcc) is of uncertain significance.
The AUG is the initiation codon encoding a methionine amino acid at the N-terminus of the protein. (Rarely, GUG is used as an initiation codon, but methionine is still the first amino acid as it is the met-tRNA in the initiation complex that binds to the mRNA). Variation within the Kozak sequence alters the “strength” of the translational start site. Kozak sequence strength refers to the favorability of initiation, affecting how much protein is synthesized from a given mRNA.

Figure 11.25 The Kozak Sequence. The sequence schematic above shows the most conserved bases around the initiation codon from over 10,000 human mRNAs. Larger letters indicate a higher frequency of incorporation. Note the larger size of A and G at the 8 position (−3, Kozak position) and at the G at position 14 which corresponds to (+4) position in the Kozak sequence.
Figure from: Audrey Michel
{kind=link}
Back to the Top
11.6 The Elongation Phase of Translation
Both prokaryotic and eukaryotic elongation phases of transcription utilize similar elongation factors during the process. Table 11.2 provides a summary of their functions.
Table 11.2 Comparison of Prokaryotic and Eukaryotic Translation Elongation Factors
![]()
Prokaryotic Elongation
The prokaryotic elongation phase of transcription requires the activity of three primary elongation factors (EFs), EF-Tu, EF-Ts, and EF-G. During elongation, aminoacyl-tRNAs are delivered to the ribosome in the form of a ternary complex: the tRNA, a translational GTPase (in bacteria: EF-Tu or SelB), and a GTP molecule (Fig. 11.26). The tRNA decodes the information on the mRNA by forming hydrogen bonds (H-bonds) between codon and anticodon nucleobases. Remarkably, the free-energy difference between correct (cognate) and incorrect (near-cognate, non-cognate) base pairing alone does not explain the very high fidelity of decoding. Rather, high fidelity is achieved by a two-step decoding process: initial selection leading to GTPase activation and proofreading. In addition to the free-energy difference, kinetic effects contribute to the discrimination. The GTP hydrolysis rate is increased and tRNA rejection rate is decreased by the recognition of the correct codon.
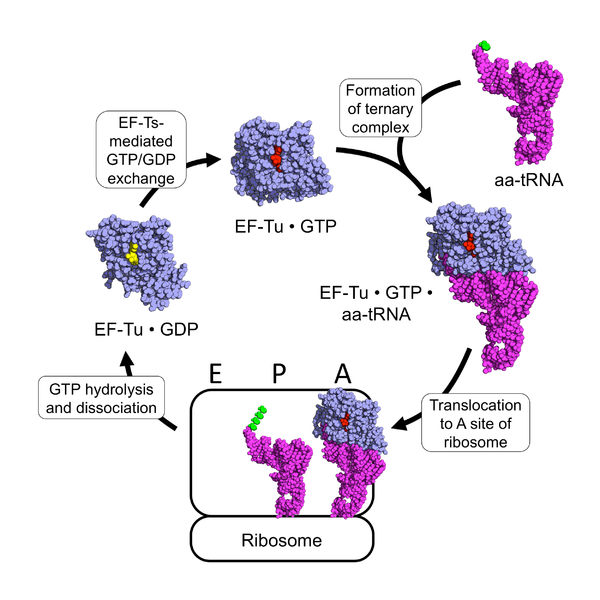
Figure 11.26 The EF-Tu tRNA Chaperone Protein. Ef-Tu (shown in blue) is a GTPase enzyme chaperone that when bound to GTP (shown in red), can form a ternary complex with an amino acid-containing tRNA molecule (tRNA shown in purple; amino acid shown in green). The ternary complex translocates to the A-site of the ribosome where the anticodon of the incoming tRNA hybridizes with the codon of the mRNA. An exact match of the codon-anticodon hybridization leads to the hydrolysis of GTP by EF-Tu and its subsequent dissociation from the ribosome (the hydrolysis product GDP is shown in yellow). EF-Tu is unable to release the GDP on its own. It requires an protein-mediated exchange of the GDP for a molecule of GTP using the elongation factor, EF-Ts.
Figure from: Awchen
{kind=link}
Small-subunit nucleotides A1492 and A1493 adopt a flipped-out conformation in the presence of a tRNA and, in this conformation, the tRNA anticodon hydrogen bonds with the codon of the mRNA forming a mini-helix structure (Fig. 11.27). The flipped out nucleotides A1492, A1493 along with G530 were found to shield the codon–anticodon base pairs from solvent. This shielding of near-cognate base pairs from the solvent is incomplete causing an increase in the free-energy difference between near-cognate and cognate base pairs and more flexibility within the docking region. This reduces the strength of hydrogen bonding between a non-cognate tRNA and causes the inappropriate tRNA to leave the A-site before peptide bond formation can occur. This increases the fidelity and discrimination of tRNA selection, such that only the correct cognate tRNA is incorporated into the A-site.
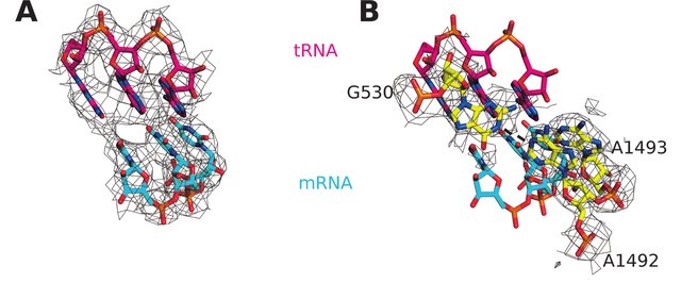
Figure 11.27 Cognate Interaction of tRNA and mRNA in E. coli. (A) Interaction of cognate tRNA anticodon (shown in red) with the mRNA codon (shown in blue) when wild type EF-Tu binds and chaperones the tRNA complex to the A-site of the ribosome. (B) Same figure as shown in (A), with the ribosomal nucleotides A1492 and A1493 depicted in the flipped out conformation.
Figure from: Fislage, M., et. al. (2018) Nuc. Acids Res. 46(11):5861-5874.
Interestingly aminoglycosides, a class of antibiotics, bind to the decoding center and lock nucleotides A1492/A1493 in the flipped-out conformation (Fig. 11.28). In this way aminoglycosides promote the accommodation of near-cognate, thus wrong, tRNAs into proteins during synthesis causing wide-spread mutagenesis. This is toxic to the bacteria and leads to bacterial cell death.

Figure 11.28 Structure of the Aminoglycoside Antibiotic, Puromycin.
Figure from: Yikrazuul
{kind=link}
After GTP hydrolysis, the GTPase EF-Tu dissociates from the tRNA. At this point, the EF-Tu is tightly bound with a molecule of GDP and cannot release GDP on its own to be recycled for a second round of tRNA chaperoning. The recharging of EF-Tu is executed by the Elongation Factor Thermo stable (EF-Ts) (Fig 11.29). The binding of EF-Ts with EF-Tu-GDP causes a conformational change in EF-Tu that allows the release of GDP. The binding of a new molecule of GTP with the EF-Tu protein causes the dissociation of EF-Ts and fully recharges EF-Tu.
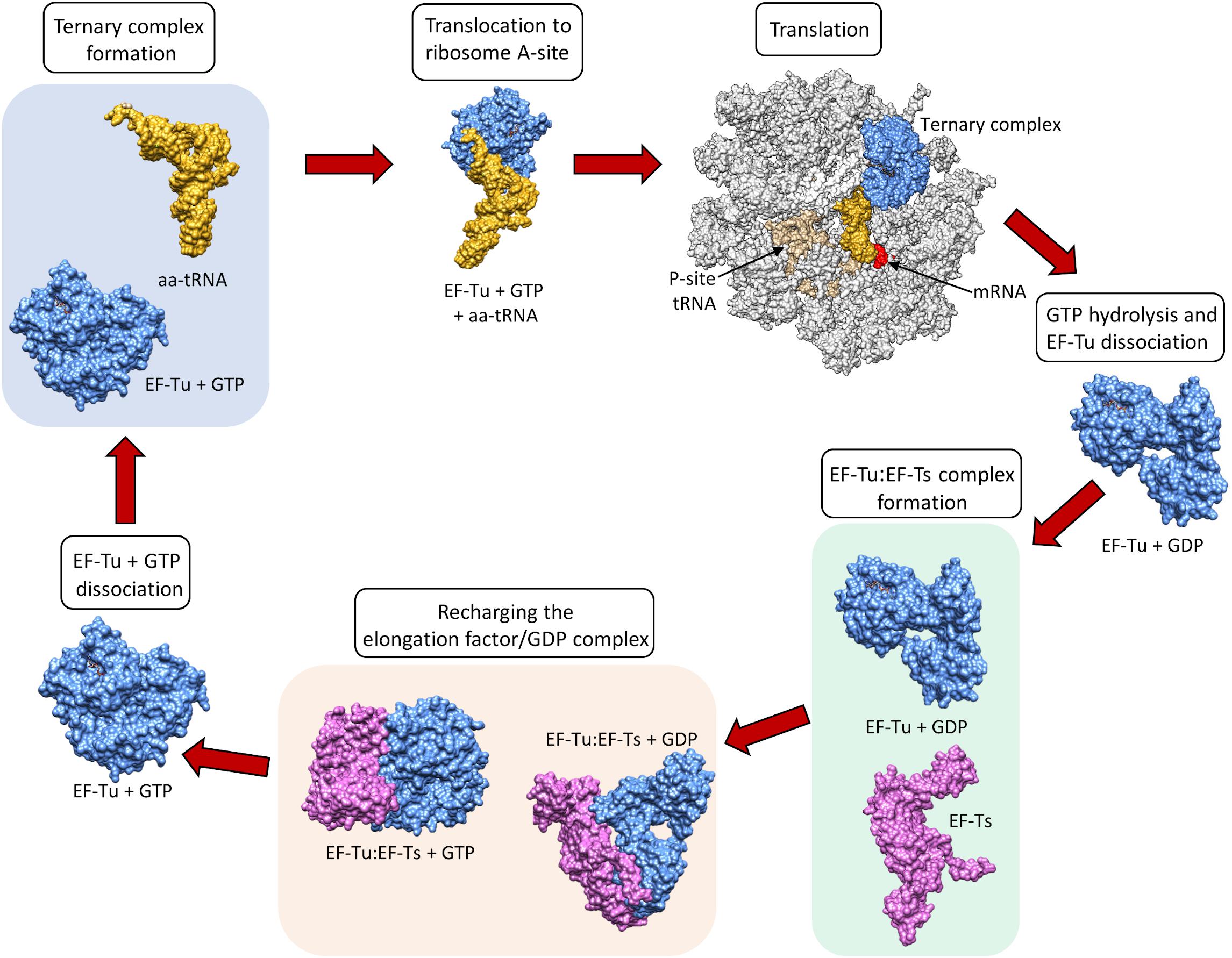
Figure 11.29 Recharging of EF-Tu is Mediated by EF-Ts. When EF-Tu is bound with GTP (shown in blue), it can bind an aa-tRNA complex (shown in yellow) causing the translocation of the aa-tRNA to the A-site of the ribosome (shown in grey). The anticodon of the tRNA is positioned in the A-site, such that it can form H-bonds with the codon of the mRNA (shown in red). Binding of the cognate aa-tRNA in the A-site causes GTP hydrolysis by EF-Tu and subsequent dissociation of EF-Tu (GDP) from the ribosome. Following the ribosomal dissociation, EF-Tu(GDP) binds with a molecule of EF-Ts (shown in purple). Binding of EF-Ts causes a conformational change in EF-Tu, such that GDP is released. A molecule of GTP is then incorporated into EF-Tu while it is complexed with EF-Ts. This causes the dissociation of EF-Ts from the complex and the EF-Tu is recharged.
Figure from: Harvey, K.L, et. al. (2019) Front. Microbiol. 10:2351
Dissociation of EF-Tu from the ribosome allows the tRNA to move into to the peptidyl transferase center (A-site) on the large subunit. At the core of ribosomal translation is the catalysis of peptide bond formation (Fig. 11.30). The current reaction models point to a substrate assisted mechanism. Simulations indicate that the transition state forms due to extensive hydrogen bonding with water molecules and the surrounding rRNA bases and that the C-O bond cleavage takes place after C-N bond formation. Peptide bond formation results in the transition of the amino acid docked on the P-site tRNA to the nascent growing peptide that is now held on the tRNA in the A-site (Fig 11.30). Note that this mechanism causes the nascent growing peptide to always grow in the N- to C- direction.
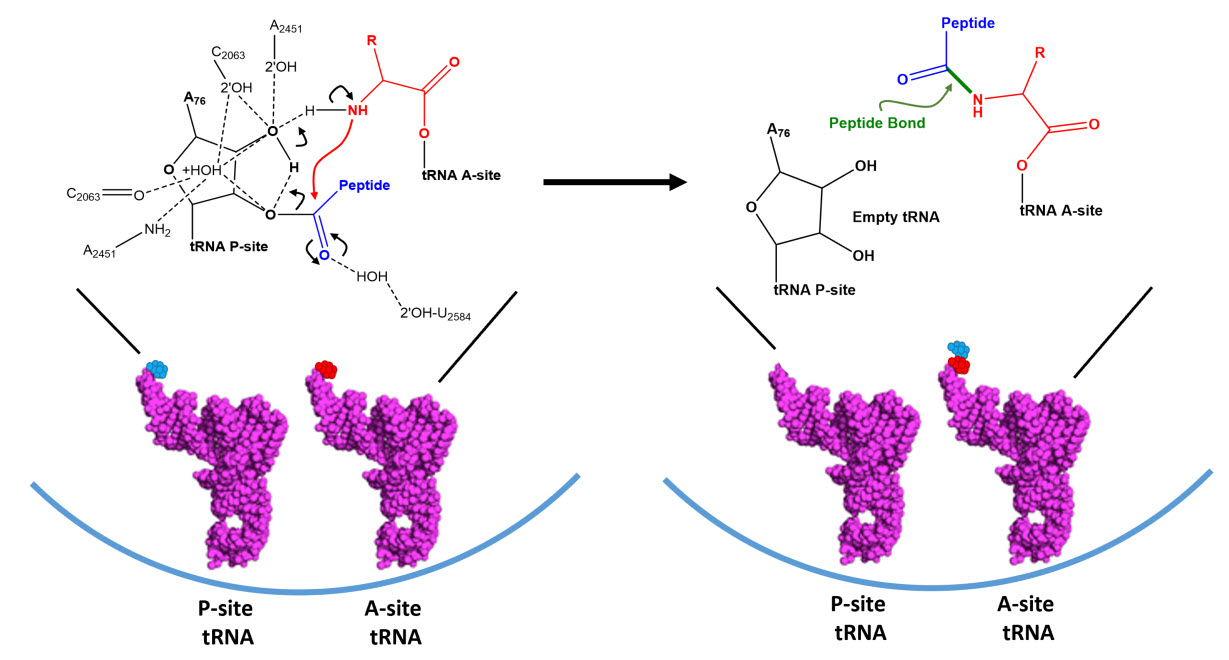
Figure 11.30 Proposed Mechanism of Peptide Bond Formation in the Ribosome. (Left) tRNA molecules docked in the P- and A-sites are oriented such that the two amino acids move in close proximity. Hydrogen bond formation with the ribosomal rRNA and surrounding water molecules stabilizes the formation of the transition state and enables the amine nigtrogen from the A-site amino acid to attack the carbonyl carbon of the P-site amino acid. Formation of the peptide bond occurs prior to the C-O bond breakage forming a transient oxyanion intermediate. (Right) The products of the peptidyl transferase reaction include the nascent growing peptide attached to the A-site tRNA and an empty tRNA in the P-site.
Figure modified from: Awchen
Once the peptide bond is formed, the ribosome needs to translocate down the mRNA to make the next mRNA codon available within the A-site. This also requires the shifting of the tRNA molecules, such that the tRNA in the A-site (which is now tethered to the nascent peptide) shifts to the P-site. The P-site tRNA (which is now empty) shifts to the E-site, and if there was an empty tRNA in the E-site, it will shift to exit the ribosome. Shifting the tRNAs and mRNAs within the ribosome core requires the action of the EF-G elongation factor (Fig 11.31).
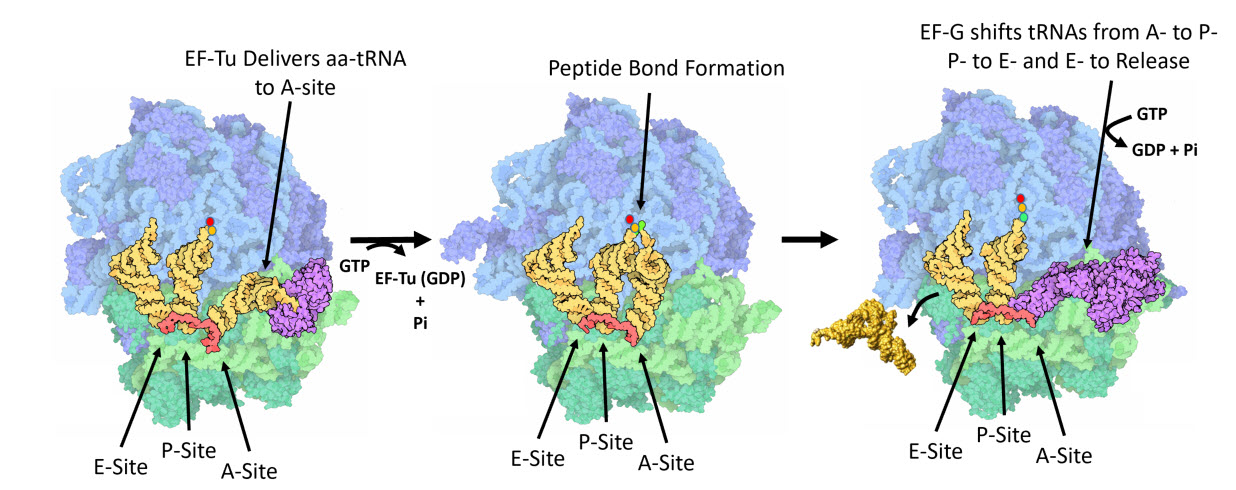
Figure 11.31 One Cycle of Elongation. (Left) During one round of amino acid elongation on a nascent peptide, the EF-Tu protein binds with the cognate aa-tRNA molecule and shuttles it to the A-site of the ribosome. GTP hydrolysis by EF-Tu leads to the hybridization of the anticodon of the tRNA with the codon of the mRNA and causes the dissoication of the EF-Tu (GDP-bound) from the ribosome. (Center) Following the dissociation of EF-Tu, the peptide bond is formed leading to the transfer of the nascent peptide from the tRNA in the P-site, to the tRNA in the A-site. (Right) Peptide bond formation leads to a conformational change in the ribosome that allows the binding of EF-G (GTP Bound) near the A-site of the ribosome. Rapid hydrolysis of GTP by EF-G causes a large conformational shift in the protein that twists the large subunit of the ribosome and shifts the bound tRNAs from the A- to the P-site; from the P- to the E-site; or from the E-site to exiting the ribosome.
Figure modified from: Goodsell, D. (2010) Molecule of the Month
{kind=link}
EF-G is a GTP hydrolase protein that binds to the A-site of the ribosome. The EF-G protein has high flexibility that enables it to act as a hinge. Folding of EF-G is dependent on GTP hydrolysis. Thus, when binding to the ribosome, the fast hydrolysis of GTP acts as a power stroke folding the EF-G protein and causing a conformation shift in the ribosome that enables the translocation of the tRNA residues and the mRNA. Translocation of tRNAs is accompanied by large-scale collective motions of the ribosome: relative rotation of ribosomal subunits and L1-stalk motion (Fig. 11.32). The L1 stalk, which is a flexible part of the large subunit, is in contact and moves along with the tRNA from the P to the E site. Once in the EF-G-GDP form, the factor quickly dissociates from the ribosome, opening up the A-site for the recruitment of the next aa-tRNA molecule. The elongation cycle will continue to be repeated until a termination codon is reached.
![]()
Figure 11.32 Large-scale Motion of the Large Subunit of the Ribosome During Translocation. (a) Pre-translocation structure of the ribosome with tRNAs in A and P sites (green, brown). The L1 stalk of the large subunit is shown in purple. (b) Motions accompanying tRNA translocation.
Figure from: Bock, L.V., Kolár, M.H., Grubmüller, H. (2018) Cur. Op. Struc. Bio. 49:27-35.
Eukaryotic Elongation
The elongation phase in eukaryotic translation is very similar to prokaryotic elongation. Essentially, the mRNA is decoded by the ribosome in a process that requires selection of each aminoacyl-transfer RNA (aa-tRNA), which is dictated by the mRNA codon in the ribosome acceptor (A) site, peptide bond formation and movement of both tRNAs and the mRNA through the ribosome (Fig. 11.33) A new amino acid is incorporated into a nascent peptide at a rate of approximately one every sixth of a second. The first step of this process requires guanosine triphosphate (GTP)-bound eukaryotic elongation factor 1A (eEF1α) to recruit an aa-tRNA to the aminoacyl (A) site, which has an anticodon loop cognate to the codon sequence of the mRNA. The anticodon of this sampling tRNA does not initially base-pair with the A-site codon. Instead, the tRNA dynamically remodels to generate a codon-anticodon helix, which stabilises the binding of the tRNA-eEF1α-GTP complex to the ribosome A site. This helical structure is energetically favourable for cognate or correct pairing, and so discriminates between the non-cognate or unpaired and single mismatched or near-cognate species. This is important for the accuracy of decoding since it provides a mechanism to reject a non-cognate tRNA that carries an inappropriate amino acid. The pairing of the tRNA and codon induces GTP hydrolysis by eEF1α, which is then evicted from the A site. In parallel with this process, the ribosome undergoes a conformational change that stimulates contact between the 3′ end of the aa-tRNA in the A site and the tRNA carrying the polypeptide chain in the peptidyl (P) site. The shift in position of the two tRNAs [A to the P site and P to the exit (E) site] results in ribosome-catalysed peptide bond formation and the transfer of the polypeptide to the aa-tRNA, thus extending the polypeptide by one amino acid. The second stage of the elongation cycle requires a GTPase, eukaryotic elongation factor 2 (eEF2), which enters the A-site and, through the hydrolysis of GTP, induces a change in the ribosome conformation. This stimulates ribosome translocation to allow the next aa-tRNA to enter the A-site, thus starting a new cycle of elongation.

Figure 11.33 Eukartyotic Translation Elongation Phase. This schematic represents the four basic steps of eukaryotic translation elongation. The ribosome contains three tRNA-binding sites: the aminoacyl (A), peptidyl (P) and exit (E) sites. In the first step of peptide elongation, the tRNA, which is in a complex with eIF1 and GTP and contains the cognate anticodon to the mRNA coding sequence, enters the A site. Recognition of the tRNA leads to the hydrolysis of GTP and eviction of eEF1 from the A site. In parallel, the deacylated tRNA in the E site is ejected. The A site and the P site tRNAs interact, which allows ribosome-catalysed peptide bond formation to take place. This involves the transfer of the polypeptide to the aa-tRNA, thus extending the nascent polypeptide by one amino acid. eIF5A allosterically assists in the formation of certain peptide bonds, e.g. proline-proline. eEF2 then enters the A site and, through the hydrolysis of GTP, induces a change in the ribosome conformation and stimulates translocation. The ribosome is then in a correct conformation to accept the next aa-tRNA and commence another cycle of elongation.
Figure from: Knight, J.R.P., et. al. (2020) Disease Models & Mechanisms 13, dmm043208.
Back to the Top
11.7 Translation Termination
Prokaryotic Termination
Termination of bacterial protein synthesis occurs when a stop codon is presented in the ribosomal A-site and is recognized by a class I release factor, RF1 or RF2. These release factors (RFs) have different but overlapping specificities, where RF1 reads UAA and UAG and RF2 reads UAA and UGA, with strong discrimination against sense codons. The RFs are multi-domain proteins, where binding and stop codon recognition by domain 2 at the decoding site causes the universally conserved GGQ motif of domain 3 to insert into the A-site of the PTC, some 80 Å away from the decoding site. This event triggers hydrolysis of the peptidyl-tRNA bond in the P-site of the PTC, and the nascent peptide chain can then be released via the ribosomal exit tunnel (Fig. 11.34). After peptide release, RF1 and RF2 dissociate from the post-termination complex. The dissociation is accelerated by a class II release factor called RF3, which functions as a translational GTPase that binds and hydrolyses GTP in the course of termination.
While RF3 increases the efficiency of peptide hydrolysis, it is not an essential protein for the process. In gene knockout studies, RF3 is dispensable for growth of Escherichia coli, and its expression is not conserved in all bacterial lineages. For example, RF3 is not present in the thermophilic model organisms of the Thermus and Thermatoga genera and in infectious Chlamydiales and Spirochaetae. This means that both RF1 and RF2 are capable of performing a complete round of termination independently of RF3 or that other GTPases from the elongation or initiation phases of translation can compensate for the action of RF3.
The release factors RF1 and RF2 acquire an open conformation (Fig. 11.34) on the 70S ribosome, which is distinctly different from the closed conformation observed in crystal structures of free RFs. The conformational equilibrium of the free RFs in solution shows that this open conformation is dominating at about 80%.
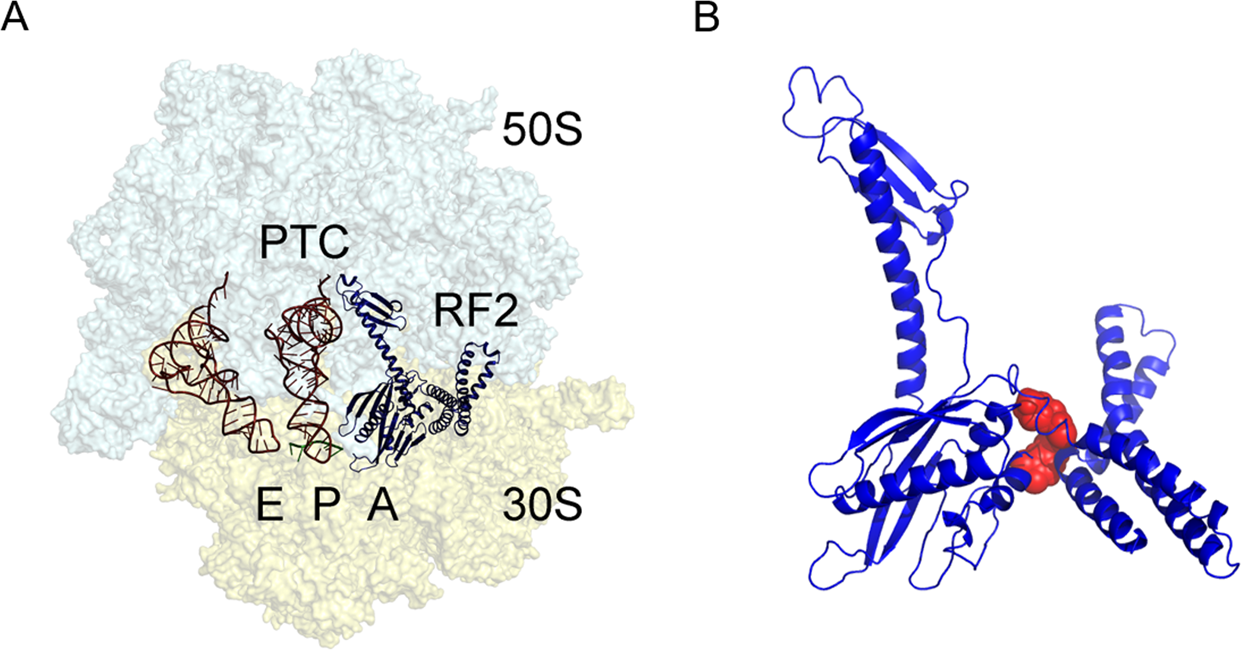
Figure 11.34. The bacterial 70S ribosome termination complex with RF2. (A) View of the ribosome termination complex with E- and P-site tRNAs (brown), mRNA (green) and RF2 (dark blue). (B) Close-up view of the hinge region of RF2 between domains 1 and 4 used for virtual screening, where the putative binding region is indicated by a docked ligand (red).
Figure from: Ge, X., et. al. (2019) Scientific Reports 9:15424.
During peptide hydrolysis, the RF factors cause rotational and conformational changes within the ribosome that allow the binding of a ribosome recycling factor (RRF) and the EF-G GTPase, which leads to the dissociation of the large subunit from the small subunit and the release of the mRNA (Fig. 11.35).
![]()
Figure 11.35 Termination of Translation. When a stop codon enters the A-site of the ribosome RF1 or RF2 enter the A-site and bind with the mRNA. This leads to the hydrolysis of the protein and release through the exit tunnel. Binding of RF3 and GTP hydrolysis causes the dissociation of the RF factors and conformational change of the ribosome structure. Subsequent binding of the ribosome recyclying factor, RRF, and EF-G causes the dissociation of the large and small ribosomal subunits and the release of the mRNA.
Figure modified from: Bock, L.V., Kolár, M.H., Grubmüller, H. (2018) Cur. Op. Struc. Bio. 49:27-35.
Eukaryotic Termination
In eukaryotes and archaea, on the other hand a single omnipotent RF reads all three stop codons. Although the mechanism of translation termination is basically the same, there is neither sequence nor structural homology between the bacterial RFs and the eukaryotic eRF1, apart from the universally conserved GGQ motif which is required for peptide hydrolysis from the tRNA. the eRF3 GTPase coordinates the release of eRF1 following hydrolysis. In Archae, there is no eRF3 homolog, instead the aEF1A protein mediates this function. The process of eukaryotic ribosomal disassembly and recycling is currently not well understood, but appears to involve an ABC type ATPase called ABCE1.
Mitochondria have independent RFs that can recognize standard and non-standard stop codons, as indicated in Figure 11.5, and are more homologous with bacterial systems of ribosomal recycling and disassembly.
Summary of Translation
An overall summary of prokaryotic translation is given in Figure 11.36.

Figure 11.36 Summary of Prokaryotic Translation. (left) Structure of the bacterial ribosome in complex with EF-Tu (PDB 5AFI). (right) Scheme of the bacterial translation cycle. 30S: small subunit; 50S: large subunit; IF1, IF2, IF3: initiation factors; fM-tRNA: N-formylmethionine tRNA; aa-tRNA: aminoacyl tRNA; EF-Tu, EF-G: elongation factors; RF1, RF2, RF3: release factors; RRF: ribosome recycling factor; green trace: nascent protein. The question mark stands for a stop codon recognition.
Figure from: Bock, L.V., Kolár, M.H., Grubmüller, H. (2018) Cur. Op. Struc. Bio. 49:27-35.
Back to the Top
11.8 Regulation of Translation
Heterogeneity of Ribosome Structure
Over the years, many studies performed in eukaryotes presented evidence that ribosomes can vary in their protein and rRNA complement between different cell types and developmental states. These observations culminated in the postulation of the ‘ribosome filter hypothesis’ by Mauro and Edelman in the year 2002. The authors propose that the ribosome composition functions as translation determination factor. Depending on the RPs and rRNA sequences represented in the respective ribosome, the complex acts like a filter that selects for specific mRNAs and hence modulates translation (Fig. 11.37). RP heterogeneity can arise from differential expression of paralogs/homologs of RP proteins within different cell types or occur due to differential post-translational modifications of RPs, such as phosphorylation. The protein to rRNA ratio may also slightly vary within ribosomal composition affecting translation efficiency and selectivity.
rRNA genes are also present in multiple copies throughout the genomes of organisms from all domains of life. For example, the bacteria Streptomyces coelicolor harbors six copies of divergent large subunit (LSU) rRNA genes that constitute at least five different LSU rRNA species in a cell. These genes were shown to be differentially transcribed during the morphological development of the organism. Similarly, B. subtilis harbors ten rRNA operons and their reduction to one copy increased the doubling time as well as the sporulation frequency and the motility of the resulting mutant.
Modification of the rRNA also provides another avenue of ribosomal heterogeneity. Similar to tRNA, rRNA residues can be chemically modified and commonly have 2-OH methylation. The conversion of uridine to psuedoruridine is also quite common. In eukaryotes, the modifications are facilitated by snoRNAs and their tissue specific expression might be a source for ribosome specialization. In light of the increasing evidence, ribosome heterogeneity, though still far from being entirely understood, proves to be an integral mechanism to modulate and fine tune protein synthesis in response to environmental signals in all organisms.

Figure 11.37. Components of the translation machinery that have the potential to contribute to functional heterogeneity. Structures were visualized with Polyview 3D software using the following maps: E. coli large ribosomal subunit (LSU; pdb accession code: 3d5a, rRNA in light gray, proteins in dark gray); small ribosomal subunit (SSU; pdb accession code: 3d5b); E. coli IF2 (pdb accession code: 1g7r); human eIF3 (EMD-2166); yeast tRNAPhe (pdb accession code: 6tna).
Figure from: Sauert, M., Temmel, H., and Moll, I. (2015) Biochimie 114: 39-47.
Effects of Sequence and Secondary Structure in mRNA
The amount of protein produced from any given mRNA (i.e., the translational output) is influenced by multiple factors specified by the primary nucleotide sequence. These factors include GC content, codon usage, codon pairs, and secondary structure.
For example, 5’UTR sequences in the mRNA may interact with small miRNAs and lead to RNA interference. miRNA interactions may also target mRNA for degradation (Figure 11.38). This process is aided by protein chaperones called argonautes. This antisense-based process involves steps that first process the miRNA so that it can base-pair with a region of its target mRNAs. Once the base pairing occurs, other proteins direct the mRNA to be destroyed by nucleases. Fire and Mello were awarded the 2006 Nobel Prize in Physiology or Medicine for this discovery.
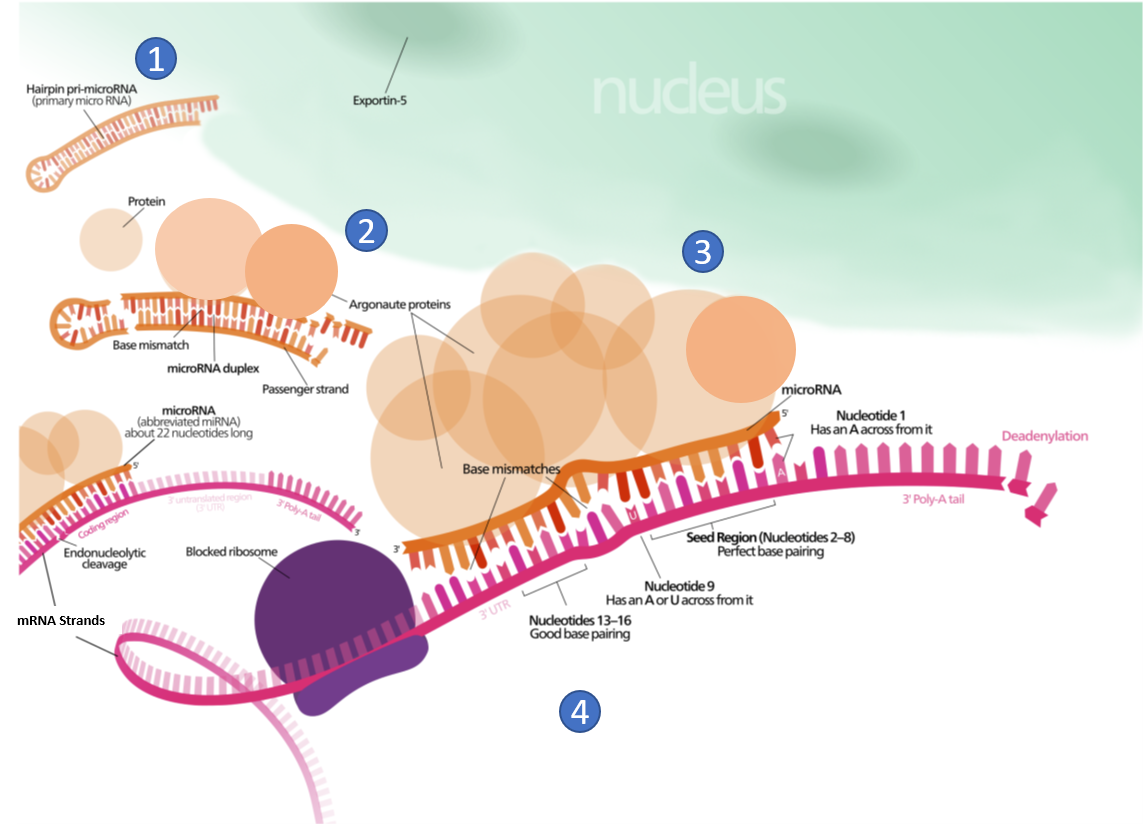
Figure 11.38 Role of Micro RNA (miRNA) in the Inhibition of Eukaryotic mRNA Translation. (1) A protein called Exportin-5 transports a hairpin primary micro RNA (pri-miRNA) out of the nucleus and into the cytoplasm. (2) An enzyme called Dicer (not shown), trims the pri-miRNA and removes the hairpin loop. A group of proteins, known as Argonautes, form a miRNA/protein complex. (3) miRNA/protein complex hydrogen bonds with mRNA based on complimentary sequence homology, and blocks translation. (4) The miRNA/protein complex binding speeds up the breakdown of the polyA tail of the mRNA, causing the mRNA to be degraded sooner.
Figure modified from: Wikimedia Commons
{kind=link}
Effects of the Nascent Peptide on Ribosome Efficiency
Since the Peptidyl Transferase Center (PTC) is buried within the large subunit, during translation the nascent peptide chain (NC) exits through a 100 Å long tunnel (Figure 11.39). The exit tunnel plays an active role in protein synthesis. Certain peptide sequences specifically interact with tunnel walls and induce ribosome stalling. Furthermore, the exit tunnel is a binding site for a clinically important class of antibiotics known as the macrolides.

Figure 11.39 The Ribosomal Exit Tunnel (a) Scheme of the ribosome exit tunnel with several proteins highlighted. NC, nascent peptide chain; ERY, erythromycin; PTC, peptidyl transferase center. (b) Context of the erythromycin (ERY, in green) binding; figure based on PDB: 5JTE. Several large subunit nucleotides are highlighted in bold red. Two proteins uL4 and uL22 form a constriction site. The nascent peptide is shown as transparent surface structure.
Figure from: Bock, L.V., Kolár, M.H., Grubmüller, H. (2018) Cur. Op. Struc. Bio. 49:27-35.
When synthesizing proteins containing proline stretches (i.e. several prolines in a row), ribosomes become stalled. Stalling is alleviated by a specialized elongation factor, EF-P in bacteria. Recently, cryo-EM structures of a ribosome stalled by a proline stretch with and without EF-P were resolved. In simulations of the PTC region, elongation factor P (EF-P) was observed to stabilize the P-site tRNA in a conformation compatible with peptide bond formation, while in the absence of EF-P, the P-site tRNA moved away from the A-site tRNA.
The exit tunnel can accommodate 30–60 AAs, depending on the level of NC compaction. The rate of translation of about 4–22 AA per second in bacteria provides the NC with sufficient time to explore its conformational space and to start folding when still bound to the ribosome-tRNA complex.
Back to the Top
11.9 References
This chapter was remixed and adapted from the following resources under creative commons licensing:
- Wikipedia contributors. (2020, June 27). Wobble base pair. In Wikipedia, The Free Encyclopedia. Retrieved 16:47, August 12, 2020, from https://en.wikipedia.org/w/index.php?title=Wobble_base_pair&oldid=964760055
- Lorenz, C., Lünse, C., and Mörl, M. (2017) tRNA modifications: Impact on structure and thermal adaptation. Biomolecules 7(2)35. Available at: https://www.mdpi.com/2218-273X/7/2/35/htm
- Pan, T. (2018) Modifications and functional genomics of human transfer RNA. Cell Research 28:395-404. Available at: https://www.nature.com/articles/s41422-018-0013-y#Sec3
- Bednárová, A., Hanna, M., Durham, I., Van Cleave, T., England, A., Chaudhuri, A., and Krishnan, N. (2017) Lost in translation: Defects in transfer RNA modifications and neurological disorders. Front. Mol Neurosci. 10:135. Available at: https://www.researchgate.net/publication/316440980_Lost_in_Translation_Defects_in_Transfer_RNA_Modifications_and_Neurological_Disorders
- Wikipedia contributors. (2020, May 17). Transfer RNA. In Wikipedia, The Free Encyclopedia. Retrieved 23:03, August 15, 2020, from https://en.wikipedia.org/w/index.php?title=Transfer_RNA&oldid=957227343
- Gomez, M.A.R., and Ibba M. (2020) Aminoacyl-tRNA Synthetases. RNA, Available at: https://rnajournal.cshlp.org/content/early/2020/04/17/rna.071720.119.abstract
- Li, R., Macnamara, L.M., Leuchter, J.D., Alexander, R.W., and Cho, S.S. (2015) MD Simulations of tRNA and Aminoacyl-tRNA Syntetases: Dynamics, Folding, Binding, and Allostery. Int. J. Mol Sci. 16(7):15872-15902. Available at: https://www.mdpi.com/1422-0067/16/7/15872
- Wikipedia contributors. (2020, August 15). Ribosome. In Wikipedia, The Free Encyclopedia. Retrieved 05:22, August 16, 2020, from https://en.wikipedia.org/w/index.php?title=Ribosome&oldid=973144885
- Kater, L., Thoms, M., Barrio-Garcia, C., Cheng, J., Ismail, S., Ahmed, Y.L., Bange, G., Kressler, D., Berninghausen, O., Sinning, I., Hurt, E., and Beckmann, R. (2017) Visualizing the assembly pathway of nucleolar Pre-60S Ribosomes. Cell 171(7):1599-1610. Available at: https://www.sciencedirect.com/science/article/pii/S0092867417314290
- Bock, L.V., Kolár, M.H., Grubmüller, H. (2018) Molecular simulations of the ribosome and associated translation factors. Cur. Op. Struc. Bio. 49:27-35. Available at: https://www.sciencedirect.com/science/article/pii/S0959440X1730132X
- Doris, S.M., Smith, D.R., Beamesderfer, J.N., Raphael, B.J., Nathanson, J.A., and Gerbi, S.A. (2015) Universal and domain-specific sequences in 23S-28S ribosomal RNA identified by computational phylogenetics. RNA 21:1719-1730. Available at: https://www.researchgate.net/publication/281141702_Universal_and_domain-specific_sequences_in_23S-28S_ribosomal_RNA_identified_by_computational_phylogenetics
- Aleksashin, M.A., Leppik, M., Hochenberry, A.J., Klepacki, D., Vázquez-Laslop, N., Jewett, M.C., Remme, J., and Mankin A.S. (2019) Assembly and functionality of the ribosome with tethered subunits. Nature Communications 10:930. Available at: https://www.nature.com/articles/s41467-019-08892-w#rightslink
- Wikipedia contributors. (2020, July 3). Formylation. In Wikipedia, The Free Encyclopedia. Retrieved 22:03, August 16, 2020, from https://en.wikipedia.org/w/index.php?title=Formylation&oldid=965827476
- Gualerzi, C.O., and Pon C.L. (2015) Initiation of mRNA translation in bacteria: structural and dynamic aspects. Cell Mol Life Sci. 72:4341-4367. Available at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4611024/
- Wikipedia contributors. (2020, June 14). Kozak consensus sequence. In Wikipedia, The Free Encyclopedia. Retrieved 06:03, August 18, 2020, from https://en.wikipedia.org/w/index.php?title=Kozak_consensus_sequence&oldid=962444929
- Knight, J.R.P., Garland, G., Pöyry, T., Mead, E. Vlahov, N., Sfakianos, A., Frosso, S., De-Lima-Hedayioglu, F., Mallucci, G.R., von der Haar, T., Smales, C.M., Sansom, O.J., and Willis, A.E. (2020) Control of translation elongation in health and disease. Dis. Mod. and Mech. 13: dmm043208. Available at: https://dmm.biologists.org/content/13/3/dmm043208
- Adio, S., Sharma, H., Senyushkina, T., Karki, P., Maracci, C., Wohlgemuth, I., Holtkamp, W., Peske, R., and Rodina, M.V. (2018) Dynamics of ribosomes and release factors during translation termination in E. coli. eLife 7:e34253. Available at: https://elifesciences.org/articles/34252
- Ge, X., Oliveira, A., Hjort, K., Bergfors, T., Guitiérrez-de-Terán, H., Andersson, D.I., Sanyal, S., and Åqvist, J. (2019) Inhibition of translation termination by small molecules targeting ribosomal release factors. Scientific Reports 9: 15424. Available at: https://www.nature.com/articles/s41598-019-51977-1#rightslink
- Svidritskiy, E., Demo G., Loveland A.B., Xu, C., and Korosteleve, A.A. (2019) Extensive ribosome and RF2 rearrangements during translation termination. eLife 8:e46850. Available at: https://elifesciences.org/articles/46850
- Sauert, M., Temmel, H., and Moll, I. (2015) Heterogeneity of the translational machinery: Variations on a common theme. Biochimie 114:39-47. Available at: https://www.sciencedirect.com/science/article/pii/S0300908414003952